а. Аномалия Киари (Chiari)
Старое название аномалия Арнольда-Киари. Это патологическое состояние, связанное с неправильным формированием структур ЗЧЯ и, в ряде случаев, ствола мозга. На настоящий момент известны четыре разновидности этой патологии. Наибольшую клиническую значимость из них представляют Chiari-1 и Chiari-2.
CHIARI-1 – характеризуется опущением миндаликов мозжечка ниже уровня большого затылочного отверстия, что часто сопровождается формированием сирингомиелии на шейно-грудном уровне.
Эпидемиология: средний возраст клинического проявления заболевания 40 лет, имеется незначительная предрасположенность женского пола (м/ж=1/1.3)
Основные симптомы: головные боли затылочного характера, боли по задней поверхности шеи, мозжечковая симптоматика (нарушения координации, шаткость походки), при наличии сирингомиелии характерными являются диссоциированные расстройства чувствительности на верхней половине туловища по типу “накидки”. В редких случаях Киари-1 может сопровождаться окклюзионной гидроцефалией. Кроме того, следует иметь в виду, что иногда у этих больных случаются ночные остановки дыхания.
Диагностика: На настоящий момент МРТ головного мозга является методом выбора при диагностике данной патологии. Следует отметить, что кроме МРТ головного мозга при подозрении на аномалию Киари-1 необходимо сделать МРТ шейного и грудного отделов спинного мозга для исключения/подтверждения сирингомиелии. И, наоборот, при наличии сирингомиелии в обязательном порядке необходимо сделать МРТ головного мозга с особым вниманием на состояние структур ЗЧЯ.
Лечение: Асимптоматичные случаи лечения не требуют. Рекомендуется лишь контролирование ситуации путем проведения ежегодного МРТ обследования. При наличии симптоматики лечение только хирургическое. Операцией выбора является “декомпрессия области краниоцервикального перехода” с пластикой оболочки.
Исходы: Улучшение состояния наблюдается у 87% оперированных больных, стабилизация – у остальных. Риск развития серьезных осложнений составляет менее 0.5%.
CHIARI-2 – часто сопутствует миеломенингоцеле. При этом патологическом состоянии отмечается еще большее опущение миндаликов мозжечка (они обычно находятся вне полости черепа), каудальная дислокация структур ЗЧЯ – т.е. ствол, 4-й желудочек расположены значительно ниже, частично или полностью в спинномозговом канале, а каудальные черепно-мозговые нервы имеют восходящее направление (в отличие от “нисходящего” в норме).
Эпидемиология: в связи с более грубой патологией и большим вовлечением в процесс ствола мозга, заболевание проявляется в детском (часто в раннем) возрасте. Чем в более раннем возрасте происходит появление симптоматики, тем серьезнее подлежащая патология и тем хуже прогноз. У новорожденных часто бывает быстрое нарастание стволовой симптоматики с летальным исходом.
Основные симптомы: связаны с нарушением функция ствола мозга (главным образом продолговатого мозга и моста) – нарушения глотания, остановки дыхания, стридор, аспирация, тетрапарез, парез мимической мускулатуры, нарушения крика (у новорожденных)
Диагностика: МРТ является методом выбора. Кроме дистопии структур ЗЧЯ часто встречаются различные другие аномалии развития головного мозга, гидроцефалия (значительно чаще, чем при Киари-1). Ларингоскопия – помогает оценить степень нарушения иннервации глотки и гортани.
Лечение: Только хирургическое. Первым этапом необходимо разрешить проблему с гидроцефалией (шунт), далее, при сохраняющейся стволовой симптоматике производится декомпрессия ЗЧЯ и области краниоцервикального перехода с пластикой оболочки. При наличии грубых бульбарных нарушений рекомендуется трахеостомия.
Исходы: Улучшение состояния наблюдается у 80% оперированных больных (у 60% после операции наблюдается практически норма), однако у оставшихся 20% часто идет прогрессирование стволовых нарушений. Наиболее неблагоприятные исходы наблюдаются у детей до года. Основная причина летальности – остановка дыхания.
CHIARI-3 – наиболее тяжелая форма дистопии, часто имеется субокципитальное энцефаломенингоцеле. Обычно эта клиническая ситуация несовместима с жизнью.
CHIARI-4 – церебеллярная гипоплазия без дистопии миндаликов.
b. Spina bifida (незаращение дужек позвоночника)
Это общее название различных типов нарушения закладки и развития нервной трубки на спинальном уровне. Выделяют две принципиальных нозологии: Spina bifida occulta –врожденное отсутствие остистых отростков нескольких позвонков и незаращение дужек без явной патологии формирования самой нервной трубки; Spina bifida aperta – представляет собой две патологических ситуации – менингоцеле (в мешке из оболочек нет нервной ткани) и миеломенингоцеле (отмечается также неправильное развитие спинного мозга и корешков).
SPINA BIFIDA OCCULTA
Общая информация: данная патология встречается примерно у 20% людей европеоидной расы и в большинстве случаев не требует никакого лечения. Однако иногда она сочетается с патологией, которая приводит к появлению и прогрессированию симптомов поражения поясничного отдела позвоночника, корешков и конуса спинного мозга. Эта патология: диастематомиелия, “фиксированный спинной мозг” (“tethetred cord”), спинальная липома или дермоид. Поэтому, любой человек с диагнозом “spina bifida” должен быть обследован с помощью МРТ спинного мозга (поясничный отдел). В случаях появления: слабость в ногах, проблемы с мочеиспусканием, деформации стоп, нарушения походки; необходима консультация нейрохирурга.
“Фиксированный спинной мозг” (“tethetred cord”) – это состояние, проявляющееся аномально низким расположением конуса спинного мозга (ниже L2 позвонка) и чаще всего связано с короткой и толстой конечной нитью (“filum terminale”), которая как бы натягивает спинной мозг и вызывает в нем нарушения кровообращения. К аналогичному патологическому состоянию могут привести спинальная липома, дермоид и диастематомиелия (все они являются “ассоциированным” комплексом spina bifida occulta).
Признаки и симптомы:
| Признак | Встречаемость (%) |
Кожные проявления (всего) гипертрихоз (излишнее оволосение) подкожная липома (без роста в спинальный канал) прочие (гемангиома, дермальный синус и т.д.) |
54% 22% 15% 17% |
| Нарушения походки за счет слабости в ногах | 93% |
| Мышечная атрофия, деформация стоп, укорочение ног | 63% |
| Чувствительные нарушения | 70% |
| Нарушения мочеиспускания | 40% |
| Нарушения мочеиспускания (как единственный дефицит) | 4% |
| Боль в пояснице, в ногах, в стопах | 37% |
| Сколиоз или кифоз | 29% |
| Spina bifida occulta (поясничная или крестцовая) | 98% |
Диагностика: на этапе предоперационного планирования всем больным в обязательном порядке проводится МРТ поясничного отдела позвоночника, у больных с нарушениями функции мочевого пузыря рекомендуется проведение цистометрографии (для оценки предоперационной функции).
Лечение: существуют две принципиальные опции: наблюдение (у пациентов без неврологической симптоматики) и хирургическое лечение – “освобождение спинного мозга” (“untethering”). Суть операции заключается в удалении существующей патологии (костного шипа, липомы, дермоида, иссечение миеломенингоцеле) и иссечении фрагмента конечной нити (“filum terminale”) для снятия натяжения со спинного мозга.
Исходы: Основной целью операции является предотвращение дальнейшего прогрессирования симптоматики (эффективность более чем 95%). Имеющиеся же симптомы в большинстве случаев остаются на предоперационном уровне. Улучшение наблюдается не более чем у 20% больных. Поэтому очень важно, чтобы при появлении первых же симптомов больной был консультирован нейрохирургом.
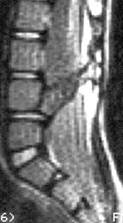
“Расщепленный спинной мозг” – выделяют два варианта этой патологии: диастематомиелия – когда костный “шип” (см.МРТ изображение) разделяет как спинной мозг, так и дуральный мешок на два отдельных “рукава”; дипломиелия – когда сам спинной мозг разделен на две половинки фиброзным тяжом и, все это находится в едином дуральном мешке. Симптоматика, показания к операции и результаты аналогичны описанным выше в разделе “Фиксированный спинной мозг” (“tethetred cord”). Операция состоит из “удаления шипа” и освобождения спинного мозга (“untethering”) путем иссечения конечной нити.
SPINA BIFIDA APERTA
Миеломенингоцеле(ММ) – врожденный дефект формирования позвоночника, оболочек и самого спинного мозга и его корешков. Внешне выглядит как выбухающий на спине новорожденного (чаще в поясничной области) “мешок” покрытый истонченным эпидермисом.
Эпидемиология: встречаемость составляет 1-2 на 1000 родившихся живых новорожденных (0.1-0.2%). Риск увеличивается до 2-3%, если в семье уже есть ребенок с ММ. Риск так же выше в семьях, где у ближайших родственников уже есть дети с ММ (особенно с материнской стороны). Наследование идет по неменделевскому типу и, возможно, мультифакториально.
Симптомы: кроме имеющегося “мешка” на спине у новорожденного может быть в той или иной степени выраженный неврологический дефицит со стороны нижних конечностей и сфинктеров. Дефицит зависит главным образом от “тяжести” патологии и от ее локализации. В общем, чем выше уровень поражения спинного мозга, тем грубее неврологический дефицит.
Диагностика: диагноз не вызывает никаких сомнений сразу по рождении ребенка. Неврологический статус оценивается ориентировочно на уровне “есть движения”/параплегия, в связи с невозможностью детального неврологического обследования у новорожденного. Особое внимание уделяется сопутствующим проблемам, т.к. у этих детей нередки различные аномалии по органам и системам (особое внимание обращается на зрелость легочного дерева при планировании ранней операции). Из дополнительной патологии ЦНС наиболее часто встречается гидроцефалия (65-85%) и аномалия Киари-2. Использование дополнительных визуализационных методик (КТ, МРТ) на дооперационном этапе не требуется.
Лечение: Все случаи ММ требуют хирургического лечения. Основная цель операции – “послойное закрытие врожденного дефекта с формированием нервной трубки”. Как только появляются признаки гидроцефалии, она должна быть контролирована с помощью шунта (часто это приходится делать одновременно с пластической операцией). Цель лечения – предотвращение новых проблем у ребенка, путем коррекции существующих. Рассчитывать на улучшение предоперационного неврологического статуса не следует.
с. Энцефалоцеле
Аналогичная spina bifida по своей природе аномалия формирования черепа и головного мозга. Образуется в результате “несращения” костей черепа и бывает в виде двух форм: менингоцеле (в мешке только оболочки) и энцефалоцеле (кроме оболочек в мешке имеется и мозговая ткань).
Эпидемиология: встречается в 5 раз реже миеломенингоцеле (0.02-0.04%)
Симптомы: наличие у новорожденного мягкотканного мешка на голове (чаще всего в затылочной области).
Диагностика: кроме визуального осмотра перед планированием операции необходимо сделать МРТ головного мозга в 3-х проекциях. Это позволит визуализировать содержимое мешка (мозг, синусы, магистральные сосуды), планировать объем операции и прогнозировать исход.
Классификация: предложена в 1972 году Suwanwela& Suwanwela, в ее основу положен принцип “локализации”:
1 – затылочные Э
2 – Э свода черепа (составляют примерно 80% всех Э):
А. межлобные Э
Б. Э области переднего (большого) родничка
В. межтеменные Э
Г. височные Э
Д. Э области заднего (малого) родничка
3 - Фронто-этмоидальные (синципитальные) Э (составляют 15% всех Э), различают 3 вида:
А. назо-фронтальные Э
Б. назо-этмоидальные Э
В. назо-орбитальные Э
4 - Базальные Э (1.5%) – это единственная группа, которая может не иметь визуальных проявлений болезни и патология часто проявляется скрытой ликвореей и “криптогенными” менингитами. ИНИЭНЦЕФАЛИЯ – наиболее неблагоприятная форма базального Э. Представляет собой грубый дефект вокруг большого затылочного отверстия. Большинство младенцев с этой патологией - мертворожденные.
5 - Э области ЗЧЯ (часто содержат структуры мозжечка и 4-го желудочка) – худший прогноз
Лечение: только хирургическое. Цель операции примерно аналогична лечению миеломенингоцеле (т.е. в основном превентивная +косметическая).
Исходы: косметический результат операции достигается без особого труда. Функциональный результат зависит от самой “подлежащей” патологии головного мозга и прямопропорционален количеству мозговой ткани, включенной в мешок. Наилучшие функциональные результаты достигаются у детей с менингоцеле. Лишь 5% детей с энцефалоцеле развиваются нормально.
d. Арахноидальные кисты головного мозга
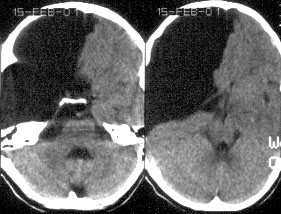
Это аномалия развития паутинной оболочки мозга, возникающая в результате неправильного расщепления ее листков в процессе формирования субарахноидального пространства (по сути, эти кисты являются “интра-арахноидальными”). Наиболее частая локализация – это область сильвиевой щели, СЧЯ, полюса височной доли. Раньше эти кисты называли “синдром агенезии височной доли”, однако, волюметрические исследования с помощью МРТ выявили, что объем мозга в обоих полушариях практически одинаков и “+объем” компенсируется дислокацией головного мозга и деформацией височной кости.
Эпидемиология: 5 на 1000 аутопсий, не связанных с патологией мозга
Симптомы: Сильно зависят от локализации кисты. В таблице ниже приведены наиболее частые симптомы в зависимости от локализации кисты:
| Кисты средней черепной ямки | Супраселлярные кисты | Распространенные супра- и инфратенториальные кисты |
Эпиприпадки Головные боли Гемипарез |
Гидроцефалия и признаки ВЧГ Краниомегалия Задержка развития Зрительные нарушения Эндокринно-обменные нарушения Симптом “неваляшки” |
Гидроцефалия и признаки ВЧГ Краниомегалия Задержка развития |
Следует отметить, что большинство супраселлярных кист и кист инфратенториальной локализации требуют хирургического лечения. Многие из кист СЧЯ являются случайными находками при МРТ или КТ, сделанными по другому поводу и, если они действительно асимптоматичны, то не требуют лечения.
Диагностика: КТ, МРТ, цистернография. Все эти методы с высокой степенью достоверности выявляют кисты. Методом же выбора на сегодняшний день следует считать МРТ, особенно при диагностике супраселлярных и инфратенториальных кист. Для супраселлярных кист обязательными являются консультации офтальмолога и эндокринолога.
Лечение: Для асимптоматичных кист вполне оправданной является опция “наблюдение” и регулярный КТ/МРТ контроль. Особо хочется отметить, что тактику ведения больного с верифицированной кистой мозга определяет нейрохирург. Лечение симптоматических кист только хирургическое и, здесь есть определенный простор для выбора наиболее оптимального метода (см. таблицу ниже).
| Тип операции | Преимущества | Недостатки |
| Разовая пункция | Просто и быстро | Часто киста реаккумулирует Высокая частота неврологических осложнений |
| Краниотомия с иссечением стенок кисты и ее сообщение с базальными цистернами | Прямая инспекция стенок кисты для исключения “опухолевой природы” Более эффективно лечатся многокамерные кисты Не требуется шунт |
Рубцово-спаечный процесс может привести к рецидиву кисты В ряде случаев ток из кисты через субарахноидальное пространство недостаточен и пациенты требуют в дальнейшем установки шунта Высокая частота осложнений (из-за резкой декомпрессии) |
| Шунтирование (чаше всего кисто-перитонеостомия) | Низкий риск осложнений самой операции Высокая эффективность лечения |
Пациент становится “шунт-зависимым” |
| Эндоскопическая фенестрация стенок кисты | Более физиологичная операция, нежели шунт Низкий риск осложнений |
Высокоэффективна лишь при кистах, имеющих отношение к желудочковой системе (супраселлярные) |
Исходы: цель операции – создать мозгу условия для нормального дальнейшего развития и функционирования. Поэтому, “уменьшение” размеров кисты – это ожидаемый положительный результат операции. Полное “исчезновение” кисты – это скорее “нежелательный” результат, т.к. высока вероятность обструкции шунта и “шунт-зависимости” пациента. Зрительные нарушения при супраселлярных кистах в большинстве случаев регрессируют, а вот эндокринопатии, в лучшем случае, остаются на дооперационном уровне.
d. Аномалия Денди-Уокера (Dandy-Walker)
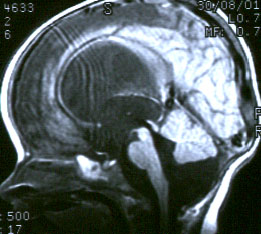
Агенезия червя мозжечка с формированием большой кисты ЗЧЯ, сообщающейся с 4-м желудочком, в результате атрезии отверстий Мажанди и Люшка. Следует четко дифференцировать это патологическое состояние от ретроцеребеллярных кист (последние редко требуют лечения и не связаны с 4-м желудочком).
Симптомы: гидроцефалия наблюдается у >90% больных с аномалией Дэнди-Уокера и, поэтому основными симптомами в раннем детском возрасте будут симптомы прогрессирующей гидроцефалии. Диагноз подтверждается путем проведения КТ или МРТ (предпочтительнее) головного мозга.
Лечение: в случае отсутствия гидроцефалии – наблюдение (редкая ситуация). При наличии водянки показана шунтирующая операция. Наиболее дискутабельным является место имплантации проксимального катетера:
в кисту (считается предпочтительным только при очень больших кистах ЗЧЯ)
в боковой желудочек (наиболее стандартная тактика)
в кисту и боковой желудочек (в случае сопутствующей окклюзии сильвиевого водопровода)
Исходы: следует иметь в виду, что аномалия Денди-Уокера является достаточно грубой патологией развития и при ней так же встречается ряд соматических проблем (главным образом аномалии сердечно-сосудистой системы). Выживаемость новорожденных в большинстве современных серий составляет >80%. Однако, функциональный неврологический исход менее благоприятен: нормальный IQ имеют лишь 50%, атаксия, спастика, нарушения координации движений являются частыми “остаточными” симптомами у этих пациентов.
Ю.В.Кушель, кмн, нейрохирург, НИИ нейрохирургии им.Бурденко
8-09-2015, 19:27