Белгородский государственный университет
Медицинский факультет
Кафедра медико-биологических дисциплин
Реферат
Наследственный микросфероцитоз.
Выполнил:
Студент 4 курса
942 группы
Базаров Виталий
Александрович
Белгород 2002
Оглавление.
Наследственный микросфероцитоз (болезнь Минковского - Шоффара)…………..2
Этиология и патогенез………………………………………………………………….2
Патологическая анатомия и патогенез………………………………………………..2
Клиническая картина…………………………………………………………………..3
Диагноз………………………………………………………………………………….4
Лечение………………………………………………………………………………….5
Наледственный микросфероцитоз (болезнь Минковского - Шоффара)
Наследственный микросфероцитоз был впервые описан в 1900 году Минковским, а в дальнейшем более подробно - Шоффаром.
Этиология и патогенез
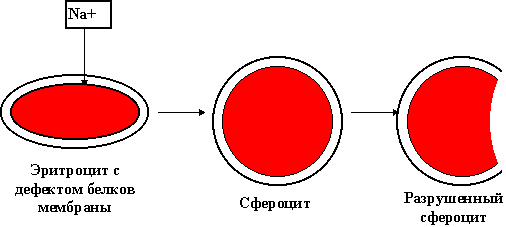
В основе заболевания лежит генетический дефект белка мембраны эритроцита. Имеющаяся аномалия мембраны приводит к проникновению в эритроцит избытка ионов натрия и повышенному накоплению в нем воды, вследствие чего образуются сферические эритроциты (сфероциты). Сфероциты, в отличие от двояковогнутых нормальных эритроцитов, не обладают способностью деформироваться в узких участках кровотока, например при переходе в синусы селезенки. Это ведет к замедлению продвижения эритроцитов в синусах селезенки, отщеплению части поверхности эритроцита с образованием микросфероцитов (отсюда название болезни - микросфероцитоз) и постепенной их гибели. Разрушенные эритроциты поглощаются макрофагами селезенки. Постоянный гемолиз эритроцитов в селезенке ведет к гиперплазии клеток ее пульпы и увеличению органа. В связи с усиленным распадом эритроцитов в сыворотке повышается содержание свободного билирубина. Поступающий в повышенном количестве в кишечник билирубин выводится из организма с мочой и главным образом с калом в виде стеркобилина. Суточное выделение стеркобилина при наследственном микросфероцитозе превышает норму в 10 - 20 раз. Следствием повышенного выделения билирубина в желчь является плейохромин желчи и образование пигментных камней в желчном пузыре и протоках.
Патологическая анатомия и патогенез
Кожа и внутренние органы при наследственном микросфероцитозе бледны и желтушны. Костный мозг в плоских и трубчатых костях гиперплазирован за счет эритроидного ростка, отмечаются явления эритрофагоцитоза ретикулярными клетками. В селезенке наблюдаются резко выраженное кровенаполнение пульпы, гиперплазия эндотелия синусов, уменьшение размеров и количества фолликулов. В печени, костном мозге, лимфатических узлах нередко выявляется гемосидероз.
Клиническая картина
Клиника зависит от выраженности гемолиза. В большинстве случаев первые признаки выявляются в юношеском или зрелом возрасте. У детей болезнь обнаруживается обычно при обследовании по поводу заболевания их родственников. Жалобы вне обострения заболевания могут отсутствовать. В период обострения отмечаются слабость, головокружение, повышение температуры. Одним из основных клинических симптомов является желтуха, которая долгое время может оставаться единственным признаком заболевания. Выраженность желтухи зависит, с одной стороны, от интенсивности гемолиза, а с другой - от способности печени к конъюгированию свободного билирубина с глюкуроновой кислотой. В моче билирубин не обнаруживается, так как свободный билирубин не проходит через почки. Кал интенсивно окрашен в темно-коричневый цвет вследствие повышенного содержания стеркобилина. В связи со склонностью к камнеобразованию у больных могут наблюдаться приступы желчнокаменной болезни, нередко сопровождающиеся признаками холецистита. В случае закупорки камнем общего желчного протока возникает синдром обтурационной желтухи (значительное повышение содержания билирубина наличие желчных пигментов в моче, кожный зуд и так далее). Кардинальным симптомом наследственого микросфероцитоза является увеличение селезенки, которая обычно выступает из-под подреберья на 2 - 3 см. При длительно протекающем гемолизе наблюдается значительная спленомегалия, в связи с чем больные жалуются на тяжесть в левом подреберье. Печень при неосложненном заболевании обычно нормальных размеров, но иногда у больных, длительно страдающих гемолитической анемией, обнаруживается ее увеличение. Могут наблюдаться признаки замедленного развития, а также нарушения лицевого скелета в виде "башенного черепа", седловидного носа, высокого стояния неба, нарушения расположения зубов, узких глазниц. Выраженность анемического синдрома различна. Часто отмечается умеренное снижение гемоглобина. У некоторых больных анемия вообще отсутствует. Наиболее резкая анемизация наблюдается в период гемолитических кризов. У лиц среднего и пожилого возраста иногда встречаются плохо поддающиеся лечению трофические язвы голени, связанные с агглютинацией и распадом эритроцитов в мелких капиллярах конечностей. Течение заболевания характеризуется так называемыми гемолитическими кризами, проявляющимися резким усилением симптомов на фоне непрерывно текущего гемолиза. При этом повышается температура в связи с массовым распадом эритроцитов, увеличивается интенсивность желтухи, появляется сильные боли в животе, рвота. Гемолитические кризы возникают обычно после интеркуррентных инфекций, переохлаждения, у женщин в связи с беременностью. Частота кризов различна, у ряда больных они не возникают.
Диагноз
Анемия при наследственном микросфероцитозе носит нормохромный характер. В мазке крови преобладают микросфероциты, отличающиеся отсутствием характерного для нормальных эритроцитов центрального просветления. Преобладание микроцитов выявляется графически на кривой Прайс-Джонса, отражающей количественные соотношения эритроцитов различных диаметров (средний диаметр нормального эритроцита составляет 7 - 7,5 мкм). При наследственном микросфероцитозе вершина кривой Прайс-Джонса растянута и сдвинута влево в сторону микроцитов. Количество ретикулоцитов увеличено. Число лейкоцитов обычно нормально. При гемолитических кризах отмечается нейтрофильный лейкоцитоз со сдвигом влево. Количество тромбоцитов варьирует в пределах нормы. В костном мозге отмечается выраженная гиперплазия эритроидного ростка. Содержание непрямого билирубина в крови повышено умеренно и, как правило, не превышает 50 - 70 мкмоль/л. Определяется повышенное содержание уробилина в моче и стеркобилина в кале. Диагноз наследственного микросфероцитоза ставится на основании течения заболевания (чередование кризов и ремиссий), клинической картины (желтуха, спленомегалия, боли в правом подреберье, анемия), данных исследования периферической крови (нормохромная анемия, ретикулоцитоз, микросфероцитоз). Важное значение имеет обследование родственников больных, у которых могут определяться едва уловимые признаки гемолиза или микросфероцитоз без клинических проявлений. Дополнительными диагностическими критериями может служить ряд лабораторных тестов. Характерным лабораторным признаком заболевания является снижение осмотической резистентности эритроцитов по отношению к гипотоническим растворам хлористого натрия. Начало гемолиза при наследственном микросфероцитозе соответствует 0,6 - 0,7 %, а конец - 0,4 % вместо 0,48 и 0,22 % в норме. Снижение осмотической резистентности свидетельствует о преобладании в крови эритроцитов сферической формы - сфероцитов, которые, менее стойки к осмотическому гемолизу, чем нормальные макропланоциты. Эритроциты больных наследственным микросфероцитозом легко разрушаются после суточной инкубации дефибринированной крови в термостате при 37° С. Добавление к эритроцитам глюкозы значительно уменьшает гемолиз, в то время как АТФ не влияет на него. Дифференциальная диагностика наследственного микросфероцитоза сводится прежде всего к диагностике гемолитических анемий вообще и требует исключения целого ряда заболеваний (аутоиммунная гемолитическая анемия, наследственный микросфероцитоз, дефицит глюкозо-6-фосфатдегидрогеназы, болезнь Маркиафавы - Микели, талассемия). Гемолитический криз, сопровождающийся анемией, лейкоцитозом с выраженным левым сдвигом, появлением в крови нормоцитов, гиперплазией эритроидного ростка костного мозга при незначительном увеличении недифференцированных клеток, наряду с увеличением селезенки, нередко дает повод к ошибочной диагностике некоторых форм лейкозов, в частности острого эритромиелоза. При дифференциальной диагностике наследственного микросфероцитоза с другими гемолитическими анемиями необходимо исключить аутоиммунные гемолитические анемии. Правильному диагнозу способствует проба Кумбса, выявляющая фиксированные на эритроцитах аутоантитела при аутоиммунных гемолитических анемиях.
Лечение
Единственным методом лечения больных наследственным микросфероцитозом является спленэктомия, которая оказывается эффективной в 100 % случаях. После спленэктомии у больных наступает практическое излечение, несмотря на то, что эритроциты сохраняют свои патологические свойства (микросфероцитоз, снижение осмотической резистентности). Прекращение гемолиза после спленэктомии объясняется удалением основного плацдарма разрушения микросфероцитов. Спленэктомия показана при частых гемолитических кризах, резкой анемизации больных, инфарктах селезенки, приступах печеночной колики. При наличии соответствующих показаний в некоторых случаях одновременно со спленэктомией может быть произведена холецистэктомия. При легких компенсированных формах заболевания у взрослых показания к спленэктомии следует ограничивать. В качестве предоперационной подготовки анемизированных больных показаны переливания эритроцитарной массы. Глюкокортикоидные гормоны при наследственном микросфероцитозе неэффективны. Прогноз при наследственном микросфероцитозе относительно благоприятен. Многие больные доживают до старости. Вероятность возникновения заболевания у детей, если один из супругов болен микросфероцитозом, несколько ниже 50 %.
Наследственные гемолитические анемии, связанные с дефицитом активности ферментов
Эта неоднородная группа заболеваний обозначается также как несфероцитарные гемолитические анемии. В отличие от микросфероцитоза они характеризуются нормальной формой эритроцитов с тенденцией к макропланоцитозу, нормальной или повышенной осмотической резистентностью эритроцитов, рецессивным типом наследования, отсутствием эффекта от спленэктомии.
Этиология и патогенез
В основе патогенеза несфероцитарных гемолитических анемий лежит дефицит активности некоторых ферментов эритроцитов, в результате чего эритроциты становятся чувствительными к воздействию различных веществ растительного происхождения, лекарственных средств. Наиболее распространенной среди этой группы заболеваний является острая гемолитическая анемия, связанная с дефицитом глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ). Согласно сведениям ВОЗ, в мире насчитывается около 100 миллионов человек с дефицитом активности Г-6-ФДГ. Наиболее часто эта аномалия встречается в странах Средиземноморского побережья (Италия, Греция), в некоторых странах Латинской Америки и Африки. В СНГ дефицит Г-6-ФДГ наиболее распространен среди жителей Азербайджана. Кроме того, носительство патологического гена описано у таджиков, грузин, русских. У детей с недостаточностью Г-6-ФДГ может проявиться фавизм. Дефицит Г-6-ФДГ наследуется по рецессивному типу, сцепленному с полом, в связи с чем клинические проявления данной патологии наблюдаются преимущественно у мужчин. При низкой активности Г-6-ФДГ в эритроцитах нарушаются процессы восстановления никотинамиддинуклеотидфосфата (НАДФ) и превращения окисленного глютатиона в восстановленный, предохраняющий эритроцит от разрушающего действия потенциальных гемолитических агентов (фенилгидразин, некоторые медикаменты, бобовые и т.д.). Гемолиз происходит преимущественно внутрисосудисто. Кожа и внутренние органы желтушны. Отмечается увеличение и полнокровие печени и селезенки, умеренное увеличение и набухание почек. Микроскопически в почечных канальцах обнаруживают гемоглобинсодержащие цилиндры. В печени и селезенке наблюдается макрофагальная реакция с наличием в макрофагах гемосидерина.
Клиническая картина
Как правило, дефицит Г-6-ФДГ не проявляется клинически без воздействия различных гемолитических агентов. Спровоцировать гемолитический криз могут противомалярийные препараты, сульфаниламиды, анальгетики, некоторые химиопрепараты (фурадонин, ПАСК), витамин К, растительные продукты (бобовые, стручковые). Выраженность гемолитического процесса зависит от степени дефицита Г-6-ФДГ и от дозы принятого препарата. Гемолиз наступает не сразу, а через 2 - 3 дня после приема препаратов. В тяжелых случаях у больных появляется высокая температура резкая слабость, боли в животе и спине, обильная рвота. Отмечается выраженная одышка, сердцебиение, нередко развитие коллаптоидного состояния. Характерным симптомом является выделение темной мочи, имеющей иногда черный цвет, что связанно с внутрисосудистым распадом эритроцитов и выделением с мочой гемосидерина. В некоторых случаях вследствие закупорки почечных канальцев продуктами распада гемоглобина и резкого снижения клубочковой фильтрации возможно развитие острой почечной недостаточности. При объективном исследовании отмечается желтушная окраска кожных покровов и слизистых оболочек, увеличение селезенки, реже печени. Через неделю гемолиз прекращается, независимо от того, продолжается прием препарата или нет.
Диагноз
В течение первых двух суток гемолитического криза у больных развивается выраженная нормохромная анемия с падением гемоглобина до 30 г/л и ниже. Отмечается высокий ретикулоцитоз, наличие нормоцитов в крови. Особенностью эритроцитов является присутствие в них телец Гейнца, представляющих собой денатурированный гемоглобин и выявляющихся при суправитальной окраске. Осмотическая резистентность эритроцитов нормальная или повышена. Со стороны белой крови во время криза отмечается лейкоцитоз со сдвигом влево до миелоцитов и более молодых форм. В костном мозге наблюдается гиперплазия эритроидного ростка и явления эритрофагоцитоза. Диагноз острой гемолитической анемии, связанной с дефицитом Г-6-ФДГ, ставится на основании типичной клинико-гематологической картины острого внутрисосудистого гемолиза, связи заболевания с приемом лекарств и данных лабораторных исследований, выявляющих снижение активности Г-6-ФДГ в эритроцитах больных, а иногда их родственников. При диагностике необходимо учитывать географическую распространенность дефицита Г-6-ФДГ.
Лечение
Основным методом лечения острой гемолитической анемии при выраженном падении содержания гемоглобина являются повторные переливания свежецитратной одногруппной крови по 250 - 500 мл 1 - 2 раза в неделю внутривенные вливания больших количеств физиологического раствора или 5 % раствора глюкозы. В качестве противошоковых препаратов применяют морфин, преднизолон, промедол. Из сосудистых средств используют кордиамин, камфору. При развитии острой почечной недостаточности проводят обычный комплекс терапевтических мероприятий, при отсутствии эффекта показано проведение гемодиализа. При нетяжелых гемолитических кризах в качестве антиоксидантного препарата назначают эревит внутримышечно по 2 мл 2 раза в день. Профилактика гемолитических кризов заключается в тщательном сборе анамнеза перед назначением средств, способных спровоцировать гемолитический криз при дефиците Г-6-ФДГ. При необходимости применения этих препаратов у лиц с дефицитом Г-6-ФДГ рекомендуется использовать средства для восстановления глютатиона. С этой целью применяют ксилит в суточной дозе 30 г в комбинации с рибофлавином в дозе 0,03 г в течение 1 - 2 месяцев. Прогноз неблагоприятен при развитии анурии и почечной недостаточности. При молниеносных формах заболевания смерть наступает от шока или острой аноксии.
 |
8-09-2015, 21:04