КОМИТЕТ ОБЩЕГО И ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ
ЛЕНИНГРАДСКОЙ ОБЛАСТИ
АВТОНОМНОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ ВЫСШЕГО
ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ
ЛЕНИНГРАДСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
Им. А.С.Пушкина
Дефектологический факультет
Кафедра логопедии
Реферат по дисциплине «Клиника интеллектуальных нарушений»
На тему «Болезнь Дауна.Болезнь Шерешевского-Тернера. Болезнь Клайнфельтера»
Выполнила:студентка 3 курса
Заочного отделения
Специальность-логопедия
Владимирова Оксана Сергеевна
Проверил:преподлаватель
Ефремов К.Д.
Санкт-Петербург 2010
ПЛАН
1.Ввведение
2. Болезнь Дауна
3 Синдром Шереше́вского — Тернера
4. Синдром Клайнфельтера
5.Прогноз.Заключение
6.Список литературы
Введение
НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ У ДЕТЕЙ
В последние годы большое внимание уделяют наследственной патологии у детей. Это связано с открытием законов наследственности, выяснением ро¬ли хромосом и генов, строения ДНК, механизмов передачи генетической информации.
Наследственные заболевания могут проявляться по-разному: в виде поро¬ков развития и различных других заболеваний детского возраста. Так, наслед¬ственные нарушения обмена веществ могут быть у детей, отстающих в разви¬тии, наследственные болезни почек — у детей с проявлениями тяжелого рахита, наследственное заболевание муковисцидоз — у детей, часто болеющих пневмонией. Раннее выявление причины заболевания позволяет своевременно назначить необходимые лечебные мероприятия.
Для своевременного выявления наследственной патологии у детей откры¬ты медико-генетические консультации. Разработаны и внедряются методы ан¬тенатальной диагностики наследственных заболеваний плода у женщин с не¬благоприятным акушерским анамнезом, использование которых позволило выявить большое число наследственных заболеваний. Начали проводить мас¬совые обследования новорожденных по скрининг-программам с применением ориентировочных методов оценки обменных нарушений. Обследуют детей первых месяцев жизни с задержкой психомоторного развития. В медико-гене¬тических консультациях решают вопросы наследования и устанавливают сте¬пень риска наследственной патологии у будущего ребенка.
Носителем наследственных свойств человека является хромосомный аппа¬рат ядра клетки, который состоит из 23 пар (46) хромосом: 22 пары — аутосомы, а 23-я пара — половые хромосомы; женский пол представлен Х-хромосомами (XX), мужской — XY-хромосомами. В каждой хромосоме в линейной последовательности содержатся гены, которые являются носителями наслед¬ственных свойств и определяют особенности развития, строения и жизнедея¬тельности организма. Хромосомы управляют процессами биосинтеза белка. Нарушения в хромосомном аппарате могут происходить на молекулярном уровне (в дезоксирибонуклеиновой кислоте — ДНК) без видимых изменений в структуре хромосом (генные мутации), что приводит к патологии обмена веществ. Возможно также изменение структуры и количества хромосом (хро¬мосомные мутации), что ведет к возникновению хромосомных болезней, со¬ставляющих 1% среди всех новорожденных. Грубые хромосомные нарушения обусловливают внутриутробную гибель плода.
С аномалиями половых хромосом связаны синдромы Клайнфелтера и Тер¬нера (Шерешевского —Тернера), с аномалиями аутосом — болезнь Дауна.
БОЛЕЗНЬ ДАУНА
Дауна болезнь (J. L.Н. Down, английский врач, 1828—1896) — болезнь, обусловленная аномалией хромосомного набора (изменением числа или структуры аутосом), основными проявлениями которой являются умственная отсталость, своеобразный внешний облик больного и врожденные пороки развития. Одна из наиболее распространенных хромосомных болезней, встречается в среднем с частотой 1 на 700 новорожденных. Оба пола поражаются с одинаковой частотой.
В основе заболевания в подавляющем большинстве случаев лежит трисомия по 21-й паре хромосом, т. е. вместо двух имеются три хромосомы, в связи с чем во всех клетках содержится по 47 хромосом [кариотип 47, XX (XY), + 21]. Поскольку частота рождения детей с Д. б. резко возрастает у женщин старше 35—40 лет, полагают, что дополнительная 21-я хромосома в большинстве случаев возникает в результате нерасхождения хромосом во время созревания женской половой клетки. Примерно в 1/3 случаев Д. б. связана с нерасхождением хромосом в мужской половой клетке.
Редко (2—3% больных) находят мозаицизм хромосомного набора: одни клетки имеют нормальный набор из 46 хромосом, другие — из 47 с лишней 21-й хромосомой. В этих случаях степень проявления основных симптомов Д. б. может быть различной в зависимости от количества клеток с аномальным хромосомным набором.
В 4—5% случаев, особенно при рождении детей с Д. б. в одной семье повторно, у больных обнаруживают не трисомию, а транслокацию 21-й хромосомы на одну из хромосом группы D или g. Эта редкая форма Д. б. представляет собой наследуемую аномалию и не зависит от возраста матери, поэтому высок риск повторного рождения больного ребенка в данной семье.
Патологическая анатомия. При морфологическом исследовании нервной системы погибших больных характерны уменьшение размеров и массы головного мозга, недоразвитие лобных и других долей, слабая дифференцировка борозд и извилин мозга. В ряде случаев встречаются аномалии развития головного мозга и крупных мозговых сосудов. Гистологически выявляется нарушение дифференцировки нервных клеток и недостаточная миелинизация нервных волокон головного и спинного мозга. Внутренние органы уменьшены в размерах. Наблюдается гипоплазия желез внутренней секреции, особенно щитовидной железы, коры надпочечников и половых желез. В печени — жировая вакуолизация, фиброз. Аорта узкая, стенки ее тонкие, крупные сосуды — меньшего диаметра. Часто отмечаются врожденные пороки сердца, желудочно-кишечного тракта и других органов.
Клиническая картина. Характерен внешний облик больного: косо расположенные глазные щели (восточного типа), широкая уплощенная переносица, дополнительная кожная складка у внутреннего угла глаз (эпикант), полуоткрытый рот, деформированные ушные раковины (рис. 1), увеличенный язык с гипертрофированными сосочками и глубокими бороздами, высокое сводчатое небо; по периферии радужки часто видны белесоватые очажки; короткая шея, стопы и кисти короткие и широкие; пальцы как бы обрублены, мизинец укорочен и искривлен, имеет одну сгибательную складку вместо нормальных двух; на ладони часто обнаруживают поперечную складку и высоко расположенный добавочный трирадиус (t’’) — точку, в которой сходятся папиллярные линии трех направлений (рис. 2); на стопах увеличен промежуток между I и II пальцами. Больные с рождения отстают в росте, поздно начинают держать голову, сидеть, ходить. Поздно и в неправильном порядке прорезываются зубы. Половое развитие резко задержано. Способность к деторождению установлена в единичных случаях. У многих больных имеются врожденные пороки сердца; нередко отмечаются пороки развития различных отделов желудочно-кишечного тракта (атрезии, стенозы, мегаколон).
В сыворотке крови обнаруживают увеличение концентрации иммуноглобулина G и снижение иммуноглобулина М. Характерны снижение сопротивляемости к инфекционным болезням, склонность к заболеванию лейкозом.
Изменения нервной системы доминируют в клинической картине болезни. У большинства больных окружность головы уменьшена, череп брахицефалической формы. С первых дней жизни ребенка выявляется мышечная гипотония; рефлекс Моро отсутствует. Отмечаются косоглазие, обычно сходящееся, слабость конвергенции, асимметрия лицевой иннервации, горизонтальный нистагм. У части детей с Д. б. обнаруживаются расстройства координации, которые проявляются при выполнении локомоторных проб, тонких движений. У всех больных имеются вегетативно-эндокринные расстройства: сухость кожи, предрасположение к ожирению, дерматитам, красный стойкий дермографизм, дистрофические изменения костей. С возрастом отмечается тенденция к нормализации мышечного тонуса, улучшается координация движений.
Психические расстройства характеризуются главным образом слабоумием по типу психического недоразвития — олигофрении, которая обнаруживается уже на первом году жизни. Отмечается диффузный характер слабоумия, при котором недоразвиты не только интеллект и мышление, но и другие психические функции (восприятие, внимание, память, речь, эмоционально-волевая сфера). Наряду с этим характерно преимущественное недоразвитие наиболее дифференцированных онтогенетически молодых функций — мышления и речи при относительной сохранности эволюционно более древних элементарных функций — эмоций и инстинктов.
Психическое недоразвитие при Д. б. в большинстве случаев на уровне имбециальности или дебильности, реже наблюдается идиотия. Суждения больных примитивны, абстрактное мышление им недоступно. Речь развивается поздно, словарный запас беден, произношение с дефектами. Характерны замедление мышления, плохая переключаемость, больные легко теряются в непривычной обстановке. Внимание неустойчивое, легко отвлекаемое. Относительно хорошо развита механическая память, выражена подражательность. Эмоции мало дифференцированы, больные пассивны и несамостоятельны, отличаются повышенной внушаемостью.
По особенностям темперамента чаще встречается вариант заболевания с преобладанием эретичности (возбудимости и раздражительности в сочетании с двигательным беспокойством), реже торпидности (вялости, пассивности и психомоторной замедленности).
Диагноз обычно несложен, в большинстве случаев устанавливается уже в родильном доме. При стертых клинических признаках необходимо цитогенетическое исследование.
Лечение. Специфических методов лечения пока не существует. Однако комплексная медикаментозная терапия в сочетании с лечебной физкультурой, массажем, педагогическим воздействием, занятиями с логопедом способствует улучшению состояния больных. Применяют различные методы стимуляции психического и физического развития — препараты ноотропного ряда, анаболические стероиды, витамины, тиреоидин, глутаминовую кислоту, липоцеребрин.
Прогноз для жизни относительно благоприятный; при тяжелых врожденных пороках сердца и желудочно-кишечного тракта и развитии лейкоза неблагоприятный.
Профилактика включает медико-генетическое консультирование больного и родителей, особенно молодого возраста (возможность транслокации хромосом), для определения степени риска повторного рождения в этой семье больного ребенка.
Синдром Шереше́вского — Тернера — хромосомная болезнь, сопровождающаяся характерными аномалиями физического развития, низкорослостью и половым инфантилизмом.
[править]Основные сведения
Впервые эта болезнь как наследственная была описана в 1925 г. Н. А. Шерешевским, который считал, что она обусловлена недоразвитием половых желез и передней доли гипофиза и сочетается с врожденными пороками внутреннего развития. В 1938 г. Тернер выделил характерную для этого симптомокомплекса триаду симптомов: половой инфантилизм, кожные крыловидные складки на боковых поверхностях шеи и деформацию локтевых суставов. В России этот синдром принято называть синдромом Шерешевского — Тернера.
Четкой связи возникновения синдрома Тернера с возрастом и какими-либо заболеваниями родителей не выявлено. Однако беременности обычно осложняются токсикозом, угрозой выкидыша, а роды часто бывают преждевременными и патологическими. Особенности беременностей и родов, заканчивающихся рождением ребенка с синдромом Тернера, — следствие хромосомной патологии плода. Нарушение формирования половых желез при синдроме Тернера обусловлено отсутствием или структурными дефектами одной половой хромосомы (X-хромосомы).
У эмбриона первичные половые клетки закладываются почти в нормальном количестве, но во второй половине беременности происходит их быстрая инволюция (обратное развитие), и к моменту рождения ребенка количество фолликулов в яичнике по сравнению с нормой резко уменьшено или они полностью отсутствуют. Это приводит к выраженной недостаточности женских половых гормонов, половому недоразвитию, у большинства больных — к первичной аменорее (отсутствию менструаций) и бесплодию. Возникшие хромосомные нарушения являются причиной возникновения пороков развития. Возможно также, что сопутствующие аутосомные мутации играют определенную роль в появлении пороков развития, поскольку существуют состояния, сходные с синдромом Тернера, но без видимой хромосомной патологии и полового недоразвития.
При синдроме Тернера половые железы обычно представляют собой недифференцированные соединительнотканные тяжи, не содержащие элементов гонад. Реже встречаются рудиментыяичников и элементы яичек, а также рудименты семявыносящего протока. Другие патологические данные соответствуют особенностям клинических проявлений. Наиболее важны изменения костно-суставной системы — укорочение пястных и плюсневых костей, аплазия (отсутствие) фаланг пальцев, деформация лучезапястного сустава, остеопороз позвонков. Рентгенологически при синдроме Тернера турецкое седло и кости свода черепа обычно не изменены. Отмечаются пороки сердца и крупных сосудов (коарктация аорты, незаращение боталлова протока, незаращение межжелудочковой перегородки, сужение устья аорты), пороки развития почек. Проявляются рецессивные геныдальтонизма и других заболеваний.
Синдром Шерешевского-Тёрнера встречается много реже, чем трисомия Х, синдром Клайнфельтера (ХХУ, ХХХУ), а так же ХУУ, что указывает на наличие сильного отбора против гамет, не содержащих половых хромосом, или против зигот ХО. Это предположение подтверждается достаточно часто наблюдемой моносомией Х среди спонтанно абортированных зародышей. В связи с этим допускается, что выжившие зиготы ХО являются результатом не мейотического, а митотического нерасхождения, или утраты X-хромосомы на ранних стадиях развития. Моносомии УО у человека не обнаружено. Популяционная частота 1:1500.
Клиническая картина и диагностика
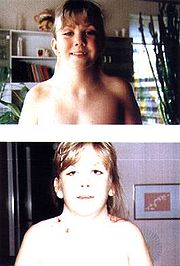
![]()
Складки кожи в области шеи — характерный признак болезни. На фото: девочка до и после пластической операции
Отставание больных с синдромом Тернера в физическом развитии заметно уже с рождения. Примерно у 15 % больных задержка наблюдается в период полового созревания. Для доношенных новорожденных характерна малая длина (42—48 см) и масса тела (2500—2800 г и менее). Характерными признаками синдрома Тернера при рождении являются избыток кожи на шее и другие пороки развития, особенно костно-суставной и сердечно-сосудистой систем, «лицо сфинкса», лимфостаз (застой лимфы, клинически проявляющийся крупными отеками). Для новорожденного характерны общее беспокойство, нарушение сосательного рефлекса, срыгивание фонтаном, рвота. В раннем возрасте у части больных отмечают задержку психического и речевого развития, что свидетельствует о патологии развития нервной системы. Наиболее характерным признаком является низкорослость. Рост больных не превышает 135—145 см, масса тела часто избыточна. У больных с синдромом Тернера патологические признаки по частоте встречаемости распределяются следующим образом: низкорослость (98 %), общая диспластичность (неправильное телосложение) (92 %), бочкообразная грудная клетка (75 %), укорочение шеи (63 %), низкий рост волос на шее (57 %), высокое «готическое» нёбо (56 %), крыловидные складки кожи в области шеи (46 %), деформация ушных раковин (46 %), укорочение метакарпальных и метатарзальных костей и аплазия фаланг (46 %), деформация локтевых суставов (36 %), множественные пигментные родинки (35 %), лимфостаз (24 %), пороки сердца и крупных сосудов (22 %), повышенное артериальное давление (17 %).
Половое недоразвитие при синдроме Тернера отличается определенным своеобразием. Нередкими признаками являются геродермия (патологическая атрофия кожи, напоминающая старческую) и мошонкообразный вид больших половых губ, высокая промежность, недоразвитие малых половых губ, девственной плевы и клитора, воронкообразный вход во влагалище. Молочные железы у большинства больных не развиты, соски низко расположены. Вторичное оволосение появляется спонтанно и бывает скудным. Матка недоразвита. Половые железы не развиты и представлены обычно соединительной тканью. При синдроме Тернера отмечается склонность к повышению артериального давления у лиц молодого возраста и к ожирению с нарушением питания тканей.
Интеллект у большинства больных с синдромом Тернера практически сохранен, однако частота олигофрении все же выше. В психическом статусе больных с синдромом Тернера главную роль играет своеобразный психический инфантилизм с эйфорией при хорошей практической приспособляемости и социальной адаптации.
Диагноз синдрома Тернера основывается на характерных клинических особенностях, определении полового хроматина (вещества клеточного ядра) и исследовании кариотипа (хромосомного набора). Дифференциальный диагноз проводится с нанизмом (карликовостью), для исключения которого проводится определение содержания гормонов гипофиза в крови, особенно гонадотропинов.
Лечение
На первом этапе терапия заключается в стимуляции роста тела анаболическими стероидами и другими анаболическими препаратами. Лечение следует проводить минимальными эффективными дозами анаболических стероидов с перерывами при регулярном гинекологическом контроле. Главным видом терапии больных является эстрогенизация (назначение женских половых гормонов), которую следует проводить с 14-16 лет. Лечение приводит к феминизации телосложения, развитию женских вторичных половых признаков, улучшает трофику (питание) половых путей, уменьшает повышенную активность гипоталамо-гипофизарной системы. Лечение следует проводить в течение всего детородного возраста больных. При синдроме Тернера у мужчин применяется заместительная терапия мужскими половыми гормонами.
Прогноз для жизни при синдроме Тернера благоприятный, исключение составляют больные с тяжелыми врожденными пороками сердца и крупных сосудов и почечной гипертензией. Лечение женскими половыми гормонами делает больных способными к семейной жизни, однако абсолютное большинство из них остаются бесплодными.
Кроме этого в последнее время проводится терапия соматотропином или человеческим гормоном роста
Синдром Клайнфельтера (0329, 0225, 0272)
Синдром Клайнфельтера встречается у 1 из 500 мальчиков. Больные с классическим вариантом синдрома имеют кариотип 47,XXY . Возможны и другие кариотипы, а у 10% больных выявляется мозаицизм 46,XY/47,XXY, встречаются и более редкие кариотипы: 48,XXXY ; 49,XXXXY ; 48,XXYY ; 49,XXXYY . Синдром обычно проявляется в подростковом возрасте как задержка полового развития. Половой член и яички уменьшены , телосложение евнухоидное , имеются гинекомастия и умеренная задержка психического развития . Больные предрасположены к сахарному диабету , заболеваниям щитовидной железы и раку молочной железы . Наличие в кариотипе не менее двух Х-хромосом и одной Y-хромосомы - самая распространенная причина первичного гипогонадизма у мужчин.
Примерно у 10% больных с синдромом Клайнфельтера наблюдается мозаицизм 46,XY/47,XXY. Поскольку в формировании фенотипа участвует клон клеток с нормальным кариотипом, больные с мозаицизмом 46,XY/47,XXY могут иметь нормально развитые половые железы и быть фертильными. Добавочная Х-хромосома в 60% случаев наследуется от матери, особенно при поздней беременности . Риск наследования отцовской Х-хромосомы не зависит от возраста отца.
Для синдрома Клайнфельтера характерен фенотипический полиморфизм. Наиболее частые признаки: высокорослость , непропорционально длинные ноги , евнухоидное телосложение , маленькие яички (длинная ось менее 2 см). Производные вольфова протока формируются нормально. В детском возрасте нарушения развития яичек незаметны и могут не выявляться даже при биопсии. Эти нарушения обнаруживают в пубертатном периоде и позднее. В типичных случаях при биопсии яичка у взрослых находят гиалиноз извитых семенных канальцев, гиперплазию клеток Лейдига , уменьшение численности или отсутствие клеток Сертоли ; сперматогенез отсутствует. Больные, как правило, бесплодны (даже если есть признаки сперматогенеза). Формирование вторичных половых признаков обычно нарушено: оволосение лица и подмышечных впадин скудное или отсутствует; наблюдается гинекомастия; отложение жира и рост волос на лобке по женскому типу. Как правило, психическое развитие задерживается, но у взрослых нарушения интеллекта незначительны. Нередко встречаются нарушения поведения, эпилептическая активность на ЭЭГ, эпилептические припадки. Сопутствующие заболевания: рак молочной железы , сахарный диабет , болезни щитовидной железы , хронические обструктивные заболевания легких .
Способы лечения бесплодия при синдроме Клайнфельтера пока не разработаны. Заместительную терапию
тестостероном обычно начинают с 11-14 лет; при
дефиците андрогенов она существенно ускоряет формирование вторичных половых признаков. У взрослых больных на фоне лечения тестостероном повышается половое влечение. При гинекомастии может
9-09-2015, 00:26