Определённую роль в процессах миграции и хоминга могут играть и хемокины. Выделены хемокины, которые повышают адгезию лимфоцитов к высокому эндотелию венул. В частности, описан цитокин из группы CC — 6CK/SLC, синтезируемый высоким эндотелием венул как периферических лимфоидных органов, так и пейеровых бляшек. Миграция T–клеток в эти лимфоидные органы резко понижена у мышей, дефектных по гену этого хемокина. Характерной особенностью действия хемокина 6CK/SLC является прекращение роллинга лимфоцитов на эндотелии, прочное прикрепление лимфоцитов к эндотелию и затем их проникновение — экстравазация. В усилении хоминга принимают участие и другие хемокины — MIP3, MIP3, SDF–1 и другие, но их усиливающее действие не является специфическим для иммунной системы ЖКТ, а общим для всей периферической иммунной системы.
Особо следует рассмотреть хемокиновый контроль привлечения в слизистую оболочку тонкого кишечника клеток B–ряда, секретирующих IgA. Эти клетки формируются в лимфатических узлах, в частности брыжеечных, и пейеровых бляшках и мигрируют в l . propria различных слизистых, в первую очередь — в слизистую тонкого кишечника. Существует два хемокина, привлекающих эти клетки. Один из них — МЕС (CCL28) — вырабатывается во всех слизистых оболочках и привлекает IgA-продуцирующие клетки, несущие на своей поверхности Рц CCR10 [355]. Другой — ТЕСК (CCL25) — специфичен для слизистой оболочки тонкого кишечника. Этот хемокин распознаётся Рц CCR9, который экспрессируется только частью IgA-пpoдуцентов, мигрирующих исключительно в тонкий кишечник. Таким образом, миграция IgA-секретирующих клеток в слизистую оболочку тонкого кишечника обеспечена более надежно, чем миграция этих клеток в другие слизистые оболочки, что и обеспечивает наибольшую концентрацию IgA–продуцентов в тонком кишечнике. Пока не выяснено, существуют ли качественные различия между клетками, реагирующими на хемотаксические сигналы, создаваемые МЕС и ТЕСК.
Роль цитокинов в функционировании иммунной системы ЖКТ.
Ранние исследования проблемы преобладающего синтеза IgA в лимфоидных структурах, ассоциированных со слизистой, показали ведущую роль T–клеток в этих процессах. Так было установлено, что клонированные T–лимфоциты из пейеровых бляшек человека и мыши индуцируют образование sIgAB–клетками, не экспрессирующими этот Рц. Эту индукцию можно воспроизвести с помощью растворимых факторов, синтезируемых T–клетками. Открытие T.R. Mosmann и R.L. Coffman [419] существования Тh1– и Тh2–клеток и различных типов цитокинов, синтезируемых этими клетками, сыграло ключевую роль в выяснении вопроса о преимущественном синтезе IgA в слизистых оболочках ЖКТ.
Продукция IgAB–лимфоцитами обеспечивается переключением генов в клетке с синтеза IgM на IgA. Это переключение происходит в B–клетках уже в пейровой бляшке и, по всей видимости, ответственными за этот процесс являются цитокины, возможно ТФР, синтезируемый Th2- или Тh3–клетками [591]. Установлено, что ТФРповышает уровень мРНК для С-области IgA в B–клетках, активированных ЛПС. Добавление ТФРк sIgA– –B–клеткам селезёнки или пейеровых бляшек, стимулированных ЛПС, селективно усиливает продукцию IgA, ИЛ–2 и ИЛ–5 существенно повышают стимулирующий эффект этого цитокина. Усиливающая роль ТФР в синтезе IgA доказана, но окончательно не установлено, какие именно факторы участвуют в переключении генов в B–клетках, локализованных в пейеровых бляшках [591]. Возможно, это межклеточные взаимодействия CD40–CD40L, а также комплекс цитокинов и биологически активных веществ кишечника типа вазоактивных пептидов, которые, как было показано, стимулируют синтез IgA в sIgA– –B–лимфоцитах [372].
В регуляции синтеза IgA в ЖКТ участвуют и другие цитокины, также синтезируемые Тh2–клетками: ИЛ–4, ИЛ–5 и ИЛ–6. Показано, что ИЛ–5 индуцирует терминальную дифференцировку sIgA+ –клеток в плазмоциты, интенсивно синтезирующие большие количества IgA. Но этот цитокин не проявляет стимулирующее действие на sIgA–B–клетки. Ещё более мощным индуктором продукции IgA является ИЛ–6. На всех sIgA+ –B–клеткax, изолированных из лимфоидной ткани кишечника человека, наблюдается высокий уровень экспрессии Рц для ИЛ–6. Добавление этого цитокина к sIgA+ –клеткам резко усиливает синтез ими IgA. Следует отметить, что sIgG+ и sIgM+ –B–клетки, выделенные из этой ткани, не содержат на поверхности Рц для ИЛ–6.В повышении синтеза IgA B–лимфоцитами играет роль синергическое взаимодействие ИЛ–6 и ИЛ–5. Предполается, что ИЛ–5 и ИЛ–6 способствуют дифференцировке B–клеток, у которых уже произошла перестройка генов в направлении синтеза IgA, но они не являются индукторами этого переключения [336].
Источником цитокинов, индуцирующих преимущественный синтез IgA, являются T–клетки фенотипа CD3,CD4 как эпителия слизистой, так и l . propria . В отличие от эпителия, среди T–лимфоцитов l . propria превалируют CD4–клетки (табл. 24, 25). С помощью техники гибридизации in situ было установлено, что среди этих клеток преобладают T–лимфоциты, экспрессирующие в основном мРНК для ИЛ–4, ИЛ–5 и ИЛ–6. Следует отметить, что клетки, экспрессирующие мРНК для ИФН — цитокина, подавляющего продукцию IgA, также выявляются в l . propria , но они преимущественно локализуются в базальном участке рядом с мышечным слоем. Как ранее отмечалось, среди эпителия слизистой преобладают T–клетки фенотипа CD3+ ,CD4– ,CD8+ , обладающие цитотоксической активностью. Но главной функциональной особенностью внутриэпителиальных лимфоцитов фенотипа CD3+ ,CD4+ ,CD8– является продукция цитокинов, способствующих дифференцировке sIgG+ –B–клеток в клетки, синтезирующие секреторный IgA. С помощью метода иммунодота было установлено, что среди внутриэпителиальных лимфоцитов фенотипа CD3+ ,CD4+ ,CD8– преобладают клетки, содержащие в цитоплазме ИЛ–4 и ИЛ–5. В то же время, было выявлено очень мало клеток, содержащих ИЛ–2 и ИФН [241, 605]. Таким образом, для T–клеток фенотипа CD3, CD4 l . propria и эпителия слизистой характерен преимущественный синтез цитокинов Th2-пpoфиля.
В целом, можно назвать по крайней мере три причины преимущественного синтеза IgAB–клетками в ЖКТ:
быстрое переключение генов с синтеза IgM на синтез IgA в активированных B–лимфоцитах пейеровых бляшек, возможно, под влиянием ТФР
способность ИЛ–5 и ИЛ–6 направлять дифференцировку B–клеток в сторону преимущественного синтеза IgA;
преобладание в l . propria кишечника Тh2–лимфоцитов, продуцирующих ИЛ–4 и ИЛ–5, которые необходимы для терминальной дифференцировки B–клеток в продуценты IgA.
Суммарное действие цитокинов, синтезируемых Th2–клетками, преимущественная локализация этих клеток в l . propria кишечника и, частично в эпителии слизистой, а также селективный хоминг в l . propria лимфоцитов, коммитированных в пейровых бляшках к синтезу IgA, и является и главной причиной преимущественного синтеза в кишечника IgA.
Эти положения о дифференцировке продуцентов IgAв известной степени относятся и к B1 –лимфоцитам, локализованным в перитонеальной полости и синтезирующим низкоавидные IgM. Экспериментально установлено, что при переносе эти клетки могут мигрировать только в перитонеальную полость и только в l . propria кишечника, но не в пейеровы бляшки (рис. 44). В кишечнике под влиянием цитокинов Тh2–клеток (ТФР, ИЛ–4, ИЛ–5) B1 –лимфоциты могут дифференцироваться в лимфоциты, синтезирующие высокоаффинные IgA, также принимающие участие в защите слизистой от инфекционных и аллергических агентов. Существует предположение, что указанные цитокины вызывают переключение генов только в активированных B1 –клетках. Иначе говоря, при проникновении в кишечник инфекционных агентов эти клетки активируются и сразу начинают продуцировать низкоаффинные IgM–АТ, составляющие первую линию обороны от инфекции, и только на эти активированные B1 –клетки действуют цитокины Тh2–клеток, вызывая переключение их генов. Вероятно, ИЛ–6 не принимает существенного участия в дифференцировке B1 –клеток, так как у мышей, дефектных по этому гену, в l . propria преобладают IgA–продуценты, экспрессирующие молекулы CD5.
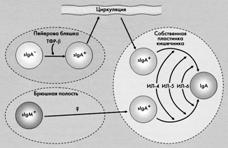
Рис. 44. Роль цитокинов в дифференцировке slgA–клеток. slgA — секреторный иммуноглобулин класса IgA; slgM — секреторный иммуноглобулин класса IgM; ТФР — трансформирующий фактор роста.
Представленные данные показывают, что B–лимфоциты в l . propria происходят из двух различных источников: пейеровых бляшек и перитонеальной полости, а цитокины, продуцируемые в этом участке кишечника, являются ответственными за их преимущественную дифференцировку в продуценты IgA.
Роль микрофлоры кишечника в функционировании иммунной системы ЖКТ и последствия ее нарушений.
Ведущую роль в функционировании как системного иммунитета, так и иммунной системы, ассоциированной с лимфоидной тканью ЖКТ, играет микрофлора кишечника. В иммунной системе новорождённых преобладает функциональная активность Тh2–клеток. Вскоре после рождения кишечник заселяется микрофлорой, которая ведёт к изменению функциональной активности T–хелперов с преобладанием таковой Тh1–клеток, характерной для взрослого организма [280, 542]. Полагают, что отсутствие или неполное переключение в онтогенезе с Тh2-активности на Тh1 является одним из факторов, определяющих развитие у детей аллергических заболеваний, в том числе и бронхиальной астмы. Одной из причин неполного переключения может быть недостаточная контаминация кишечника микроорганизмами, что является следствием существенного улучшения социально-гигиенических условий жизни [281].
Тh1–клетки являются воспалительными и их избыточная функциональная активность —частая причина возникновения различных хронических воспалительных процессов, в том числе и кишечника, в основе которых лежит ГЗТ (болезнь Крона, неспецифический язвенный колит, язвенная болезнь желудка, вызванная Helicobacterpilori, целиакия) [372]. При всех этих заболеваниях человека и у мышей с дефектными генами ИЛ–10, ИЛ–2 или избыточной продукцией TNF наблюдается преобладание в l . propria Тh1–клеток, синтезирующих ИФН и TNF. Предполагается, что мишенями для действия в l . propria цитокинов Тh1–клеток являются миофибробласты, составляющие матрикс этой структуры. Под влиянием цитокинов эти клетки выделяют фактор роста кератиноцитов, вызывающий гиперпролиферацию эпителия слизистой кишечника, и металло-протеаз, разрушающих матрикс l . propria [372]. Сочетанное действие этих биологически активных факторов лежит в основе хронических воспалительных процессов кишечника, первично инициированных Тh1–клетками.
Несмотря на то, что микробные Аг являются главными индукторами Тh1–клеток, воспалительные процессы кишечника являются относительно редким явлением. Это может быть объяснено несколькими факторами, препятствующими гиперфункции Тh1–клеток. Прежде всего это чисто механическая функция эпителия — эпителиальный барьер кишечника в нормальном состоянии непроницаем для большинства микробов. Проникновению микробов в l . propria препятствуют также гуморальные и клеточные факторы местного иммунитета. К гуморальным факторам относится секреторный IgA, который выделяется в просвет кишечника и препятствует взаимодействию аллергенов и микробных Аг со слизистой. Снижение его синтеза может вести к развитию аутоиммунных и аллергических заболеваний, что часто и наблюдается при селективном дефиците IgA. К клеточным факторам относится функциональная активность нейтрофилов l . propria , убивающих и разрушающих бактерии, проникшие в этот отдел слизистой оболочки. У детей с наследственными дефектами фагоцитоза нередко развиваются поражения кишечника, идентичные болезни Крона. Не менее значимым является упоминавшееся выше преобладание в l . propria иммуносупрессивного окружения для Th1–клеток. Наличие в этом отделе повышенного уровня ИЛ–10, ТФР, ПГЕ2 , а также малое количество АПК, ведёт к подавлению функциональной активности T–лимфоцитов и их гибели. Нарушение проницаемости слизистой, проникновение и персистенция в l . propria микробных Аг сопровождается активацией Тh1–лимфоцитов и развитием хронического воспалительного процесса.
Нарушение физиологического баланса между Тh1- и Th2–клетками в сторону повышения функций Th2 также может быть причиной заболеваний кишечника. Так, при эозинофильном гастроэнтерите, вследствие гиперфункции Тh2–клеток и повышения синтеза ими соответствующих цитокинов, происходит инфильтрация l . propria кишечника эозинофилами и тучными клетками. Пищевая энтеропатия может быть обусловлена формированием популяции ЦТЛ, специфических к определённому пищевому продукту. Одной из причин развития аллергических воспалительных процессов кишечника может быть снижение продукции цитокинов Тh1-профиля, в частности ИФН, что ведёт к повышенному образованию IgE и развитию аллергического процесса. Гиперфункция Тh2–клеток наблюдается также при паразитарных инвазиях, но, вероятно, является её следствием, а не причиной.
9-09-2015, 00:01