Министерство здравоохранения Украины
Запорожский государственный медицинский университет
Кафедра биологической химии и лабораторной диагностики
Реферат
на тему:
«Биохимия сахарного диабета»
Выполнила:
студентка 2 курса 14 группы
медицинского факультета
Чмуль Карина Олеговна
г. Запорожье, 2007 г.
План
- Инсулинзависимый сахарный диабет
- Клеточный иммунитет
- Иммунный ответ на эндогенные и эндоцитированные белки
- Интерлейкин-1
- Модель аутоиммунной гибели -клеток
- дефицит инсулина
- Коматозные состояния (острые осложнения) при диабете
- Гликирование белков - одна из главных причин поздних осложнений сахарного диабета
- Диабетические ангиопатии
- Диабетические макроангиопатии.
- Микроангиопатии.
Инсулинзависимый сахарный диабет
1. При ИЗСД происходит разрушение -клеток в результате аутоиммунной реакции
Гипергликемия и другие первичные симптомы ИЗСД обусловлены дефицитом инсулина, который в свою очередь вызван уменьшением количества -клеток (а также островков Лангерганса) в поджелудочной железе. Множество экспериментальных и клинических исследований указывает на то, что разрушение островков происходит в результате клеточной аутоиммунной реакции.
При манифестации (т.е. первом клиническом проявлении) ИЗСД почти всегда обнаруживается воспалительная реакция в поджелудочной железе - инсулит. Панкреатический инфильтрат при ИЗСД содержит Т-лимфоциты, В-лимфоциты, натуральные киллеры и макрофаги. При этом инфильтрат образуется только в тех островках, в которых есть -клетки. В островках, продуцирующих глюкагон, соматостатин, но не содержащих -клеток, нет и инфильтрата. Такая локальность, точечность реакции указывает на то, что причиной ее являются компоненты и свойства, присущие только -клеткам. Как показывают многие наблюдения, специфичность повреждения -клеток может быть следствием клеточной аутоиммунной реакции.
Клеточный иммунитет. Основными молекулами, обеспечивающими клеточный иммунитет, являются Т-рецепторы и белки главного комплекса гистосовместимости (белки ГКГ). Эти два семейства молекул принадлежат к суперсемейству иммуноглобулинов, в которое входит также семейство иммуноглобулинов (антител), давших название всему суперсемейству. В отличие от антител, которые находятся в жидкостях организма в растворенном состоянии, Т-рецепторы и белки ГКГ - это интегральные белки клеточных мембран.
Т-рецепторы имеются на поверхности Т-лимфоцитов, а белки ГКГ - на поверхности практически всех клеток. Т-рецепторы представляют собой гетеродимеры , содержащие межцепочечную дисульфидную связь. Каждая цепь содержит глобулярные вариабильный и константный домены, экспонированные на наружной поверхности мембраны, а также трансмембранный домен и короткий цитоплазматический домен:
Строение Т-рецепторов (а) и белков ГКГ классов I (б) и II (в). Стрелки указывают на пептиды - лиганды белков ГКГ; М - 2-микроглобулин |
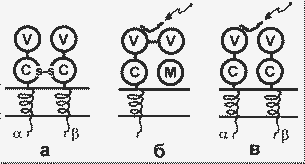
Т-рецептор составляет часть многомолекулярного белкового комплекса, включающего в общей сложности 7- 9 пронизывающих мембрану пептидных цепей. Этот комплекс формируется в цитозоле и затем включается в мембрану. Существует множество клонов Т-лимфоцитов, различающихся по структуре вариабильного домена, т.е. множество Т-рецепторов с разной специфичностью к лигандам. Разнообразие Т-рецепторов возникает так же, как и разнообразие антител, т.е. в результате соматической рекомбинации генов. Лигандами для Т-рецепторов служат короткие пептиды (10 - 20 аминокислотных остатков), которые образуются из чужеродных белков в результате протеолитической фрагментации. При этом для узнавания рецепторами необходимо, чтобы такие пептиды были соединены с белками ГКГ.
Известны два класса белков ГКГ, несколько различающихся по структуре и функциям. Белки класса I содержат две нековалентно связанные пептидные цепи - легкую и тяжелую. Тяжелая цепь своей большой N-концевой частью экспонирована на наружной поверхности клеточной мембраны, далее следуют небольшие трансмембранный и цитоплазматический домены. Легкая цепь представлена 2-микроглобулином (2m). Внеклеточная часть тяжелой цепи содержит три глобулярных домена: 1 и 2 - вариабильные домены, 3 - константный домен, сходный по структуре с пептидом 2m.
Белки ГКГ класса II - это гомодимеры; на поверхности клетки экспонированы вариабельный и константный глобулярные домены обеих цепей.
Белки ГКГ класса I имеются практически во всех клетках организма человека, а класса II - только в макрофагах, В-лимфоцитах и некоторых специализированных эпителиальных клетках. В геноме человека имеется лишь несколько генов (генных локусов) белков ГКГ (гены HLA). Однако в популяциях человека известно большое количество аллельных вариантов этих белков - варианты белков класса I и варианты белков класса II; отдельные индивиды могут наследовать лишь один (гомозиготы) или два (гетерозиготы) из этих вариантов, причем вероятность наследования разными индивидами одинаковых вариантов ничтожна. Т.о. между людьми существуют индивидуальные различия по белкам ГКГ. Именно с этим связана трансплантационная несовместимость индивидов.
Белки ГКГ являются рецепторами небольших пептидов (длиной в 10 - 20 аминокислотных остатков). Центр связывания этих пептидов образуют вариабельные домены белков ГКГ. Пептиды-лиганды могут образоваться в результате протеолитической фрагментации как собственных белков организма, так и чужеродных белков; в последнем случае пептиды-лиганды служат антигенами, вызывают иммунную реакцию с участием Т-лимфоцитов. К пептидам, образовавшимся из собственных нормальных (не мутантных) белков на ранних стадиях эмбрионального развития вырабатывается иммунологическая толерантность.
Комплекс белка ГКГ с пептидом служит лигандом Т-рецептора определенного клона Т-лимфоцитов. Т-лимфоцит своим Т-рецептором присоединяется к клетке, представившей на своей поверхности комплекс ГКГ/пептид, и если пептид в этом комплексе происходит не из собственного, а из чужеродного белка, Т-лимфоцит активируется, и включается механизм уничтожения клетки, несущей чужеродный пептид. Подчеркнем, что Т-рецептор связан не отдельно с белком ГКГ, и не отдельно с петидом-антигеном, а именно с комплексом этих молекул, которые вместе и в равной мере участвуют в образовании центра связывания для Т-рецепторов. Т.о. специфичность иммунного ответа есть результат вариабельности белков ГКГ, которые определяют и выбор пептида-антигена, и выбор Т-лимфоцита соответствующего клона.
Т-лимфоциты в организме человека представлены тремя типами: цитотоксические Т-лимфоциты (Т-киллеры), имеющие механизм уничтожения клеток, и два типа лимфоцитов, выполняющих регуляторные функции - Т-хелперы и Т-супрессоры. Т-хелперы, присоединившие антиген, стимулируют остальные компоненты иммунной системы: специфичные к данному антигену другие Т-лимфоциты, а также и В-лимфоциты. Т-супрессоры, наоборот, подавляют активность этих клеток. Т-хелперы, вероятно, играют главную роль в инициации иммунного ответа. В частности, пролиферация и окончательная дифференцировка В-лимфоцитов, узнавших чужеродный антиген, требует активации Т-лимфоцитами.
Таблица 2. Иммунный ответ на эндогенные и эндоцитированные белки
 |
Чужеродные белки могут появиться в клетке двумя путями: 1) образоваться в самой клетке (вирусные белки, мутантные белки); 2) проникнуть путем эндоцитоза в клетки макрофагов и некоторых других фагоцитирующих клеток (любые белки, появляющиеся в жидкостях организма). Ответ клеточного иммунитета в этих случаях будет несколько различным (табл. 2).
На рисунке приведена схема инициации клеточного иммунного ответа на эндоцитированный чужеродный белок:
Инициация клеточного иммунного ответа |
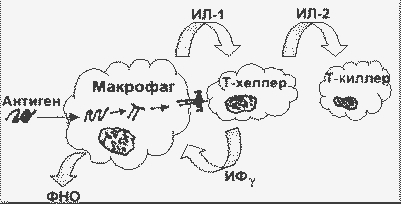
Антиген (Аг), обычно растворимый белок , часто гликопротеин, эндоцитируется антигенпредставляющими клетками (АПК; например, тканевыми макрофагами или В-лимфоцитами). В эндоцитозе участвует рецептор антигена на поверхности АПК. Комплекс Аг-рецептор интернализуется, в эндосоме происходит частичный протеолиз с образованием пептидов длиной в 10 - 20 аминокислотных остатков, пептиды соединяются с белками класса II главного комплекса гистосовместимости. Затем эндосома сливается с плазматической мембраной, и комплекс антигенный пептид/класс II-ГКГ экспонируется на поверхности клетки. Экспонированный комплекс может быть распознан Т-хелперами специфического клона, несущими подходящий Т-рецептор.
Когда Аг узнается Т-хелпером, он (Т-хелпер) активируется прежде всего в отношении транскрипции ряда цитокиновых генов. Продукция цитокинов (см. ниже) вызывает хемотаксис лейкоцитов к месту, где происходят эти события, активацию эндотелиальных клеток, пролиферацию и дифференцировку рекрутированных лейкоцитов, апоптоз и много других биологических активностей.
2. Интерлейкин-1 может быть токсичным для -клеток
В развитии клеточной аутоиммунной реакции участвуют цитокины. Это сигнальные молекулы паракринного и аутокринного действия, но некоторые из них иногда бывают и в крови в физиологически активной концентрации. Известны десятки разных цитокинов. К ним относятся интерлейкины (лимфокины и монокины), интерфероны, пептидные факторы роста, колониестимулирующие факторы. Цитокины представляют собой гликопротеины, содержащие 100 - 200 аминокислотных остатков. Большинство цитокинов образуется и действует во многих типах клеток и реагирует на разные стимулы, включая механическое повреждение, вирусную инфекцию, метаболические нарушения и др. Исключение составляют интерлейкины (ИЛ-1 и ИЛ-1) - их синтез регулируется специфическими сигналами и в небольшом количестве типов клеток.
Цитокины содержатся в тканях в пикомолярных и наномолярных концентрациях, и с высоким сродством взаимодействуют со специфическими рецепторами на наружной поверхности плазматической мембраны клеток. Цитокины участвуют в регуляции пролиферации, дифференцировки, хемотаксиса, секреции, апоптоза. ИЛ-1, фактор некроза опухолей (ФНО) и интерферон (ИФg) являются главными медиаторами развития острой фазы воспаления, инфекции и травмы. Они имеют перекрывающуюся, но все же разную биологическую активность. Клетки разных типов, или разной степени дифференцированности, или находящиеся в разном функциональном состоянии могут по-разному реагировать на один и тот же цитокин.
Цитокины действуют на клетки через специфические мембранные рецепторы и протеинкиназные каскады, в результате активируется фактор транскрипции - белок, который транслоцируется в ядро клетки, находит специфическую последовательность ДНК в промоторе гена, являющегося мишенью данного цитокина, и активирует этот ген.
В экспериментах с изолированными островками Лангерганса животных показано, что ИЛ-1 практически полностью подавляет стимулированную глюкозой секрецию инсулина и нарушает нормальную структуру островков. В островках снижается выживаемость клеток, отмечается фрагментация ДНК, уменьшается содержание ДНК, т.е. индуцируется апоптоз. При этом повреждаются преимущественно -клетки; вероятно, это связано с тем, что в островках именно -клетки имеют наибольшую плотность рецепторов ИЛ-1. Глюкоза защищает клетки от токсического действия ИЛ-1 (увеличивает выживаемость клеток). При этом индуцируется синтез белков, в частности bcl-2, ингибирующего апоптоз.
Цитокины ИФg и ФНОa усиливают токсическое действие ИЛ-1: в их присутствии ИЛ-1 токсичен для островков в гораздо меньших концентрациях. Другие цитокины не проявляют токсического действия в отношении островков.
ИЛ-1 индуцирует, в частности, синтез фермента NO-синтазы.Оксид азота NO - короткоживущий свободный радикал с высокой реакционной способностью. Он участвует в регуляции ряда физиологических функций, например, регулирует тонус сосудов (сосудорасширяющее действие), обладает противоопухолевым действием, токсичен для микроорганизмов. NO образуется при действии NO-синтазы (NOS), превращающей аргинин и кислород в цитруллин и NO. Есть два основных типа NO-синтазы: конститутивная форма (обнаружена в основном в нейронах и эндотелиальных клетках) и индуцибельная форма (iNOS) (во многих клетках, в том числе в b-клетках островков). Синтез iNOS индуцируется цитокинами и бактериальными липополисахаридами; фермент продуцирует значительно больше NO, чем конститутивные формы. Повидимому, iNOS и NO служат одним из иеханизмов защиты от микроорганизмов. NO проявляет летальное действие по отношению к простейшим, грибкам, бактериям и вирусам.
В островках Лангерганса iNOS образуется, по-видимому, только в b-клетках. В островках человека синтез мРНК и белка iNOS индуцируется при одновременном наличии двух или трех цитокинов: ИЛ-1 + ИФg или ИЛ-1 + ИФg + ФНО. В целом повреждение и гибель -клеток при аутоиммунной реакции можно представить следующим образом:
Модель аутоиммунной гибели -клеток |
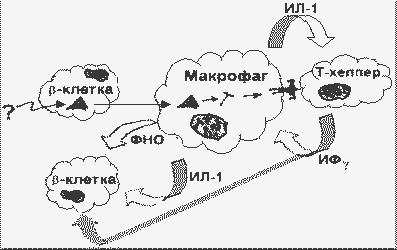
В ранней фазе иммунного ответа происходит взаимодействие одной АПК с одной Аг-узнающей клеткой. При этом повышается локальная концентрация цитокинов с паракринным действием на ближайшее окружение. Позднее развивается воспалительная реакция с участием активных иммунокомпетентных клеток, происходит секреция цитокинов, активация протеаз, образование кислородных радикалов, других иммунных медиаторов. Т.о. гибель клеток происходит, по-видимому, как по механизму некроза (воспаление), так и по механизму апоптоза.
Интерферон g (ИФg) обеспечивает положительную обратную связь с макрофагами в отношении продукции ИЛ-1 и ФНО, вследствие чего начавшийся с одной клетки иммунный ответ не затухает, а амплифицируется.
Остается неясным вопрос о природе антигена, запускающего реакцию клеточного иммунитета, избирательно направленную на -клетки. Интересные результаты получены в исследованиях на мышах линии NOD (non obesity diabetes) с высокой генетической предрасположенностью к ИЗСД. Из тканей этих мышей выделены клоны лимфоцитов, введение которых здоровым мышам вызывает диабет. Кроме того, такие лимфоциты оказались способными связывать инсулин, причем узнаваемой частью почти всегда был фрагмент -цепи, включающий 9 - 23 аминокислотные остатки (пептид В). В этих лимфоцитах пептид В соединен с белками ГКГ класса II. Авторы допускают, что инсулин может быть первичным аутоантигеном при ИЗСД у мышей NOD, а возможно и у человека.
Аутоиммунный процесс в островках поджелудочной железы развивается в течение нескольких лет и приводит к гибели основной массы (около 80%) -клеток до клинического дебюта болезни. В результате дефицита инсулина нарушается складирование энергоносителей и проявляется клиническая картина ИЗСД.
3. При дефиците инсулина нарушается синтез гликогена и жиров
При сахарном диабете инсулин-глюкагоновый индекс снижен. Это связано не только с уменьшением секреции инсулина, но и с увеличением секреции глюкагона (инсулин ингибирует секрецию глюкагона). В результате ослаблена стимуляция процессов складирования и усилена стимуляция мобилизации запасов, усилена настолько, что печень, мышцы, жировая ткань даже после приема пищи функционируют в режиме постабсорбтивного состояния. В этой драматической коллизии продукты переваривания, а также их метаболиты, вместо того, чтобы складироваться в форме гликогена и жиров, циркулируют в крови. Вероятно, в какой-то мере происходят и затратные циклические процессы типа одновременно протекающих гликолиза и глюконеогенеза, или синтеза и распада жиров и т.п..
Для всех форм сахарного диабета характерна сниженная толерантность к глюкозе, т.е. гиперглюкоземия после приема пищи или даже и натощак. При концентрации глюкозы в крови больше 180 мг/дл наступает глюкозурия. Повышена концентрация в крови липопротеинов (главным образом ЛОНП), свободных жирных кислот, кетоновых тел. В свою очередь гиперглюкоземия является основной причиной как острых, так и поздних осложнений диабета.
4. Коматозные состояния (острые осложнения) при диабете развиваются в результате нарушения обмена глюкозы и жиров
Коматозные состояния при сахарном диабете могут быть разного патогенеза. Различают три основные формы:
1. кетоацидотическая кома, с абсолютной инсулиновой недостаточностью
2. гиперосмолярная кома, с умеренной недостаточностью инсулина
3. лактатацидотическая кома, с выраженной гипоксией, сепсисом, сердечно-сосудистым шоком.
Кроме того, при инсулинотерапии сахарного диабета может быть гипогликемическая кома, связанная с передозировкой инсулина. Первые три состояния могут развиваться не только при сахарном диабете, но и при действии многих других факторов (токсических, инфекционных и др.).
Три основные формы коматозного состояния практически никогда не встречаются в чистом виде, однако обычно преобладают проявления какой-нибудь одной из форм (часто - гиперосмолярной), что и дает основания для выделения основных форм.
Первичной причиной кетоацидоза является инсулиновая недостаточность: в период комы С-пептид и иммунореактивный инсулин (ИРИ) в крови не определяются. Гипергликемия отмечается всегда, 20 - 30 ммоль/л, а иногда и более. Ацидоз при диабетической коме является следствием накопления органических кислот - кетоновых тел, а также лактата и пирувата. Концентрация кетоновых тел достигает 2 ммоль/дл (в 200 раз больше нормы); она повышается не только вследствие синтеза в печени, но и потому, что снижается экскреция кетоновых тел в связи с олигурией и анурией, которая часто бывает при коме. Снижение рН крови наблюдается всегда, до 7 и ниже (норма 7,4).
Развивается дегидратация, дефицит воды может быть до 10% от общей массы тела. Количество циркулирующей жидкости уменьшается на 25 - 30%, в результате снижается кровяное давление.
Кислородное и энергетическое голодание миокарда, уменьшение объема крови ведут к сердечно-сосудистой недостаточности. Могут быть повышение свертываемости крови, инфаркт миокарда, инфаркты паренхиматозных органов, инсульт, периферические тромбозы.
Диабетическая кома развивается медленно, в течение нескольких дней, но иногда может развиться за несколько часов. Появляются тошнота, рвота, черты лица заостряются, глаза западают, нарастает безучастность к окружающему, заторможенность, переходящая в глубокую кому (полностью выключенное сознание, отсутствие рефлексов, атония мышц и др.). В помещении, где находится больной, ощущается явственный запах ацетона. Артериальное давление снижено, почти всегда наблюдается олигурия или анурия.
Диабетическая кома требует немедленного лечения, которое включает следующие мероприятия:
1. ликвидация инсулиновой недостаточности путем введения инсулина в дозах, обеспечивающих постепенное снижение концентрации глюкозы в крови до уровня, близкого к нормальному
2. регидратацию организма путем введения жидкости
3. восстановление нормального солевого состава и рН жидкостей организма путем введения соответствующих солевых растворов
4. восстановление запасов гликогена в организме.
Проявления комы обычно ликвидируются в течение 2 - 3 дней при непрерывно продолжающемся лечении, причем лечение в первые несколько часов имеет решающее значение для спасения жизни больного.
До развития методов лечения диабета инсулином больные умирали вскоре после начала болезни от диабетической комы. Но и теперь кома наблюдается нередко. В частности, первое проявление болезни в 15 - 30% случаев сопровождается кетоацидозом и комой. Смертность от диабетической комы остается высокой - от 1 до 30%. Но основной причиной смерти больных диабетом в настоящее время являются поздние осложнения.
5. Гликирование белков - одна из главных причин поздних осложнений сахарного диабета
Поздние осложнения сахарного диабета связаны прежде всего с повреждением кровеносных сосудов (диабетические ангиопатии). Основной механизм повреждения тканей - гликирование (гликозилирование) белков, неферментативная реакция глюкозы со свободными аминогруппами белковой молекулы (Лиз, Арг, N-концевая аминокислота):
 |
При этом образуется нестабильная альдиминовая группировка, которая может превращаться в ряд других, более стабильных соединений (“ранние продукты гликозилирования”). Понятно, что при этом функции белка могут быть нарушены в результате изменения заряда белковой молекулы, ее конформации или блокирования активного центра. Глюкозилирование - медленная реакция, в тканях здоровых людей обнаруживаются лишь небольшие количества гликозилированных белков. При гипергликемии реакция существенно ускоряется. Например, у больных диабетом в состоянии гипергликемии содержание одного из гликозилированных гемоглобинов - HBA1c - в течение 2 -3 недель увеличивается в 2 - 3 раза. Степень гликозилирования разных белков неодинакова; в основном она зависит от
8-09-2015, 19:54