ГОХСПО САМАРСКИЙ МЕДИЦИНСКИЙ КОЛЛЕДЖ ИМ.
Н. ЛЯПИНОЙ
Реферат
На тему: «ХРОНИЧЕСКИЙ ЛИМФОЛЕЙКОЗ»
Выполнила: студентка 540 группы
отделения « Лабораторная диагностика»
по дисциплине
« Методы клинических исследований»
Серова Т. Г.
САМАРА 2009
Содержание:
1. Представление о хронических лейкозах
2. Хронический лимфолейкоз
3. Патогенез.
4. Клиника лимфолейкоза
5.Клинические классификации
6. Лабораторная диагностика
7. Список использованной литературы
ХРОНИЧЕСКИЕ ЛЕЙКОЗЫ
Хронические лейкозы (ХЛ) представляют собой группу опухолей кроветворной ткани, возникающих вследствие опухолевой трансформации стволовых полипотентных клеток или коммутированных клеток-предшественников. ХЛ подразделяются на две подгруппы: миелоидные и лимфоидные (миело- и лимфопролиферативные заболевания). Наиболее частой формой миелоидных опухолей является хронический миелолейкоз, при котором все клеточные линии принадлежат к опухолевому клону. Клеточные элементы при хронических миелоидных лейкозах долгое время сохраняют способность к дифференцировке и созреванию.
Хронические лейкозы лимфоидной ткани в свою очередь разделяют на две основные группы: Т- и В-клеточные. Опухолевая трансформация происходит на различных этапах дифференцировки клеток с последующим блоком в их дальнейшем развитии. Эти нарушения обычно касаются одной клеточной линии.
Течение хронических лейкозов характеризуется прогрессированием лейкемического процесса, которому сопутствует неизбежное накопление клеточной массы опухоли в костном мозге и периферической крови. Постепенное нарастание количества опухолевых элементов костного мозга приводит к вытеснению элементов нормального кроветворения, которое сначала носит относительный, а затем приобретает абсолютный характер, к замене жирового костного мозга на активный лейкозный (опухолевый). Этим изменениям гемопоэза, как правило, сопутствуют клинические симптомы, свойственные лейкозам, такие как анемия, инфекционные осложнения, геморрагический синдром. Завершается лейкоз развитием бластного криза либо гематосаркомы. Все формы хронических лейкозов сопровождаются недостаточностью костномозгового кроветворения.
Хронический лимфолейкоз
Хронический лимфолейкоз (ХЛЛ) относится к опухолям, первично возникающим в костном мозге в результате опухолевой трансформации чаще В-лимфоцитов, реже Т-лимфоцитов и последующей их моноклональной пролиферацией.
ХЛЛ впервые описан в 1845 году, а в середине 60-х гг. охарактеризован Galton и Dameshek как заболевание, сопровождающееся пролиферацией аномальных лимфоцитов. ХЛЛ составляет 30% всех регистрируемых случаев лейкозов в Европе и Америке, значительно реже выявляется в Азии. Частота встречаемости заболевания - 2,7-3,0 на 100 тысяч населения. Болеют в основном лица старше 50 лет, мужчины в два раза чаще, чем женщины. В детском и юношеском возрасте заболевание встречается крайне редко. Заболеваемость ХЛЛ наблюдается в 2 раза чаще у представителей белой расы, чем негроидной.
Большинство случаев ХЛЛ составляет В-клеточная форма (95%). На долю Т-клеточной формы приходится около 5% от всех наблюдений ХЛЛ, в основном регистрируемых в странах Азии.
Среди этиологических факторов рассматривается воздействие химических веществ, вирусов. Доказана роль человеческого Т-клеточного вируса I типа (HTLV-I) в развитии Т-клеточного варианта ХЛЛ.
Патогенез
Опухолевая трансформация происходит на уровне ранних В-лимфоцитов с последующим блоком в их дальнейшей дифференцировке и пролиферацией клона опухолевых клеток. Лимфоидные элементы при ХЛЛ как бы «заморожены» на данном этапе дифференцировки и вся лейкозная популяция является практически мономорфной. Накопление опухолевых клеток связано с нарушением процессов регуляции программированной клеточной смерти (апоптоза). В экспериментальных работах доказана сверхэкспрессия генов семейства Вс1-2 в опухолевых В-лимфоцитах, блокирующих апоптоз, что способствует удлинению продолжительности жизни этих клеток. ХЛЛ относится к медленно прогрессирующим заболеваниям. Опухоль постепенно вытесняет нормальные гемопоэтические клетки, что со временем приводит к развитию недостаточности костномозгового кроветворения. Пролиферация опухолевых В-лимфоцитов в костном мозге, лимфатических узлах, селезенке, реже в других органах (кожа, желудочно-кишечный тракт, почки, легкие и др.) обуславливает клиническую картину заболевания.
Клиника
На протяжении нескольких лет заболевание протекает бессимптомно. Лишь выявление абсолютного лимфоцитоза при исследовании клеточного состава периферической крови может привлечь внимание врача. Заболевание сопровождается общими для многих злокачественных опухолей неспецифическими симптомами - слабостью, быстрой утомляемостью, повышенным потоотделением, потерей массы тела, связанными с опухолевой интоксикацией.
По мере нарастания опухолевой массы отмечается постепенное увеличение лимфатических узлов: в первую очередь шейных и подмышечных. В последующем процесс может распространиться практически на любую группу лимфоузлов. Пораженные лимфатические узлы при ХЛЛ безболезненны, не имеют местной кожной гиперемии, эластичные, подвижные, не спаяны с кожей. Внезапный рост лимфатического узла, изменение его консистенции («деревянной плотности») требует цитологического исследования с целью возможного выявления трансформации ХЛЛ в лимфосаркому.
Увеличение лимфатических узлов при ХЛЛ обычно происходит медленно, но со временем их увеличение может приводить к сдавлению близлежащих органов и нарушению их функции. По мере прогрессирования процесса выявляется гепатоспленомегалия.
Для больных ХЛЛ характерна повышенная восприимчивость к инфекции вследствие нарушений в системе клеточного и гуморального иммунитета. Наиболее часто встречаются бактериальные и вирусные инфекции со стороны дыхательной и мочевыводящей систем, вызванные стафилококками, стрептококками, различными грамнегативными микроорганизмами. В развернутой стадии заболевания могут наблюдаться инфекции, вызванные Candida и Aspergillus, а также вирусом герпеса и цитомегаловирусом.
Дефект противоопухолевого иммунитета является причиной повышенной склонности больных ХЛЛ к развитию вторичных опухолей. Наиболее часто при ХЛЛ встречается рак кожи и кишечника.
Клинические классификации
Существуют различные клинические классификации ХЛЛ. В зависимости от локализации опухоли, особенностей течения заболевания в классификации, предложенной А.И. Воробьевым и М.Д. Бриллиант (1985 г., с изменениями и дополнениями 1999 г.), выделено несколько форм ХЛЛ - доброкачественная, прогрессирующая, опухолевая, селезеночная, абдоминальная, костномозговая. Согласно классификации, предложенной Rai К. с соавт., выделяют 5 стадий болезни (табл. 13).
Классификация ХЛЛ (Rai, 1975)
Таблица 13
| Стадия | Характеристика | Группа риска | Медиана выживаемости |
| 0 | Лимфоцитоз в крови (>15х10 /л) и костном мозге (>40%) | Низкая | Более 10 лет |
| I | Лимфоцитоз и лимфаденопатия | Промежуточная | 9 лет |
| II | Лимфоцитоз, сплено-и/или гепатомегалия, +/- лимфаденопатия | Промежуточная | 6 лет |
| III | Лимфоцитоз и анемия (НЬ<100г/л), +/- лимфаденопатия, гепатоспленомегалия | Высокая | Менее 3 лет |
| IV | Лимфоцитоз, тромбоцитопения (<100х109 /л) независимо от увеличения лимфатических узлов и органов | Высокая |
Классификация BinetJ. с соавт. предусматривает 3 стадии ХЛЛ, обозначаемые буквами А, В и С, которые коррелируют со средней продолжительностью жизни больных (табл. 14).
A. Помимо предусмотренного диагнозом лимфоцитоза крови и костного мозга, имеется одностороннее или двухстороннее увеличение лимфатических узлов в 1-2 областях, анемия и тромбоцитопения отсутствуют.
B. Увеличение лимфатических узлов в 3 областях и более. Анемия и тромбоцитопения отсутствуют.
C. Независимо от количества зон с увеличением лимфатических узлов и наличия увеличения органов имеется анемия и тромбоцитопения.
Таблица 14
Классификация ХЛЛ (BinetJ., 1981)
| Стадия | Характеристика | Выживаемость |
| А | Гемоглобин >100г/л; тромбоциты > 100,0 х 109 /л; менее 3 групп лимфатических узлов вовлечены в процесс | Более 10 лет |
| В | Гемоглобин >100г/л; тромбоциты > 100,0 х 109 /л; более 3 групп лимфатических узлов вовлечены в процесс | 5 лет |
| С | Гемоглобин <100 г/л и/или тромбоциты <100,0 х 109 /л | 2 года |
Лабораторная диагностика
В зависимости от стадии заболевания костный мозг может быть нормо- или гиперклеточным. Процент лимфоцитов в стернальном пунктате колеблется в широких пределах от 20-30% вплоть до тотальной мономорфной лимфоидной инфильтрации. По данным трепано-биопсии поражение костного мозга носит очаговый или диффузный характер. Независимо от стадии заболевания диффузная инфильтрация костного мозга лимфоидными клетками сочетается с малой продолжительностью жизни больных (менее 43 мес.) по сравнению с очаговой инфильтрацией (100 мес).
В начальной стадии заболевания картина периферической крови обычно представлена нормальным или незначительно повышенным количеством лейкоцитов. Как правило, анемия и тромбоцитопения отсутствуют. Основным гематологическим показателем при ХЛЛ является абсолютный лимфоцитоз.
Согласно рекомендациям экспертов ФАБ группы диагноз ХЛЛ считается установленным при абсолютном количестве лимфоцитов в крови, превышающем 10х103 /мкл, наличии более 30% лимфоцитов в костном мозге и иммунологическом подтверждении существования В-клеточного клона лейкемических клеток.
Указывается, что ХЛЛ должен быть заподозрен уже при абсолютном количестве лимфоцитов в крови более 5х107мкл. Другие исследователи считают, что диагноз ХЛЛ может быть поставлен при наличии абсолютного лимфоцитоза менее 5х103 /мкл, но с учетом доказательства моноклональности процесса. В лейкоцитарной формуле процент морфологически зрелых лимфоцитов составляет от 45 до 95%, встречаются единичные пролимфоциты (фото 34).
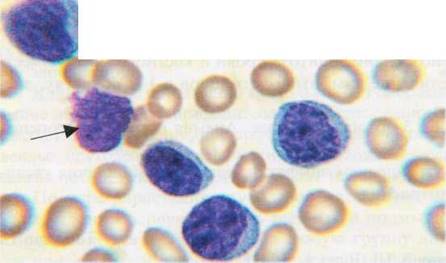
Фото 34. Периферическая кровь при хроническом лимфолейкозе.
Лимфоциты, клетка цитолиза. X900.
Имеет место относительная или абсолютная нейтропения. Лимфоциты периферической крови при ХЛЛ характеризуются небольшими размерами (7-10 мкм), округлым ядром, грубым, тяжистым распределением хроматина, отсутствием нуклеол, узкой, базофильного цвета, цитоплазмой. Встречаются клетки цитолиза. В развернутой стадии заболевания нарастает лейкоцитоз, относительный и абсолютный лимфоцитоз, нейтропения, наблюдается нормохромная анемия и/или тромбоцитопения. В лейкоцитарной формуле пролимфоциты составляют менее 10%, встречаются при просмотре препарата лимфобласты.
Обнаружение лимфоцитов с расщепленными, перекрученными, неправильной формой ядрами, грубой тяжистой или волокнистой структурой хроматина свидетельствует о возможной лейкемизации лимфосаркомы или Т-клеточном варианте ХЛЛ. В периферической крови могут обнаруживаться единичные миелоциты и метамиелоциты, обычно на фоне инфекционных заболеваний, а также нормобласты.
Примерно у 15% больных ХЛЛ наблюдается развитие аутоиммунной гемолитической анемии, реже тромбоцитопении за счет образования аутоантител к эритроцитам или эритро-кариоцитам костного мозга, тромбоцитам. Развитие анемии сопровождается раздражением красного ростка в костном мозге, ретикулоцитозом, появлением нормобластов в периферической крови, повышением содержания неконъюгированного (непрямого) билирубина в сыворотке крови. Аутоиммунный генез анемии подтверждается положительной прямой пробой Кумбса и другими тестами, выявляющими аутоантитела.
Цитограмма лимфатических узлов (ЛУ) при ХЛЛ характеризуется пролиферацией мономорфной популяции морфологически зрелых лимфоцитов. Подобная цитограмма ЛУ наблюдается при лимфоцитарной (зрелоклеточной) лимфосаркоме (лимфоме из малых лимфоцитов), но при этом, в отличие от ХЛЛ, отсутствуют изменения в периферической крови. В то же время, при лейкемизации лимфоцитарной лимфосаркомы картина крови, костного мозга и цитограмма лимфатических узлов аналогичны ХЛЛ.
Морфологический субстрат и иммунологический фенотип лимфоцитов идентичен при ХЛЛ и лимфоцитарной лимфосаркоме, поэтому высказывается точка зрения о различных проявлениях одного и того же опухолевого процесса.
Иммунологический фенотип. В 95% случаев ХЛЛ имеет В-клеточный иммунологический фенотип с экспрессией поверхностных В-клеточных антигенов CD 19, CD20, CD24, CD79a и активационных антигенов CD5, CD23, CD43. Экспрессия CD5 считается обязательной для иммунологического подтверждения В-ХЛЛ. Однако описаны редкие наблюдения ХЛЛ (7%) с отсутствием CD5 на В-лимфоцитах. CD23 используют для дифференциальной диагностики В-ХЛЛ и лейкемизации лимфомы из клеток зоны мантии лимфатического узла, имеющей аналогичный В-ХЛЛ иммунофенотип, но без экспрессии CD23. Для ХЛЛ в отличие от нормальных В-лимфоцитов и лимфосарком характерна слабая экспрессия поверхностных иммуноглобулинов (чаще slgM, реже IgM +IgD с одинаковыми легкими цепями).
Практически у всех больных наблюдается гипогаммаглобулинемия со снижением концентрации нормальных иммуноглобулинов (IgM, IgG, IgA), что отражает нарушение гуморального иммунитета и повышает чувствительность больных ХЛЛ к инфекциям. При использовании современных методов электрофореза можно обнаружить белок Бене-Джонса в моче, значительно реже встречается моноклональный иммуноглобулин в сыворотке крови.
Цитогенетика. У 50-60% больных ХЛЛ обнаруживают клональные хромосомные аберрации, наиболее часто - трисомия 12, структурные дефекты в 13, 14 хромосомах. Изучение цитогенетических особенностей клеток имеет прогностическое значение. Средняя продолжительность жизни больных с хромосомными аномалиями значительно короче (7,7 лет), чем без таковых (до15 лет).
Прогноз. ХЛЛ является достаточно медленнотекущим заболеванием. Средняя продолжительность жизни составляет около 10 лет.
Исход ХЛЛ. Наиболее часто встречается трансформация ХЛЛ в пролимфоцитарный лейкоз, что характеризуется нарастанием лейкоцитоза, числа пролимфоцитов, анемии и тромбоцитопении. Эти изменения сопровождаются резкой лимфаденопатией, спленомегалией, развитием рефрактерности к проводимой терапии.
В 3-10% случаев наблюдается синдром Рихтера (крупноклеточная анапластическая лимфосаркома). Он характеризуется ухудшением общего состояния больных, развитием общих симптомов, таких как лихорадка, потеря веса, потливость, генерализацией экстрамедуллярного опухолевого процесса с резким увеличением лимфатических узлов и/или экстранодаль-юя локализацией очагов опухолевого роста. Возможна трансформация ХЛЛ в острый лейкоз (чаще острый лимфобластный лейкоз, L2 вариант), реже в миеломную болезнь.
Список использованной литературы:
1. Руководство по гематологии. / Под редакцией А.И. Воробьева. – Москва: Медицина, 1985. – Т.1.
2. Окороков А.Н. Диагностика болезней внутренних органов. – Москва: Мед. Лит., 2001 – Т.4.
3. Дворецкий Л.И. Железодефицитные анемии. - М.:Ньюдиамед-Ао, 1998.- 37с.
4. Идельсон Л.И. Гипохромные анемии.- М.Медицина, 1981.-с.3-128
5. Справочник Видаль. Лекарственные препараты в России: Справочник М.: OVPEE-Астра Фарм Сервис, 2000 .- 1408с.
8-09-2015, 19:59