НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ОБМЕНА ВЕЩЕСТВ
К настоящему времени известно более 2500 наследственных ферментопатий, 20 классов наследственных болезней обмена, но только для части из них (для 300) установлен точный уровень метаболического блока и характер ферментативного дефекта. До сих пор большая часть этих заболеваний не диагностируется или выявляется поздно, когда происшедшие нарушения носят необратимый характер. Одна из трудностей ранней диагностики заключается в том, что в период новорожденности у этих детей нет специфических расстройств, а поздние проявления фенотипически схожи с заболеваниями ненаследственного генеза. Вторая особенность состоит в том, что для наследственных заболеваний обмена веществ характерен клинический полиморфизм, обусловленный генетической гетерогенностью. Это объясняется наличием множественных изоаллельных мутаций и возможностью возникновения мутаций в разных генах.
Клинические проявления наследственных болезней обмена веществ во многом определяются поражением нервной системы (особенно при нарушениях обмена аминокислот, липидов и кислых гликозамино-гликанов), что в свою очередь, усиливает имеющиеся нарушения и усугубляет тяжесть клинических проявлений заболевания. Для диагностики наследственных болезней важен анализ неврологических симптомов, особенно на ранних стадиях развития, и разграничение их от фенокопий- заболеваний ненаследственной природы со сходной клинической картиной. Важное практическое значение имеет выявление гетерозиготных носителей мутантного гена при наследственных болезнях. Например, при частоте фенилкетонурии 1:10 000, мутантный ген встречаетсяв популяции 1:50. Гетерозиготное состояние может клинически не обнаруживаться, поэтому выявление умеренно выраженных изменений метаболизма является единственным указанием на наличие мутантного гена.
Наследственные болезни обмена аминокислот
Роль аминокислот для организма человека чрезвычайно велика. Аминокислоты являются основными структурными элементами белков, необходимы для синтеза иммуноглобулинов, гормонов, служат источником энергии. Каждый фермент или белок имеет специфические свойства и функции, которые определяют и регулируют сложные обменные процессы и развитие организма.
Часть аминокислот не может синтезироваться в организме человека. Это незаменимые аминокислоты: триптофан, фенилаланин, метионин, лизин, лейцин, изолейцин, валин и треонин. В детском возрасте к их числу относится гистидин, т.к. организм ребенка не может синтезировать эту аминокислоту в необходимых для нормального роста количествах. Клетки растущих тканей содержат аминокислоты в высоких концентрациях, что является свидетельством высокой интенсивности процессов транспорта аминокислот через клеточные мембраны.
Для обеспечения нормального роста и развития важно не только количество поступающих аминокислот, но и их соотношение. При избытке или недостатке аминокислот развиваются явления аминокислотного дисбаланса. Например, избыток лейцина в пище тормозит рост организма, метионина- вызывает токсическое поражение нервной системы, цистина- способствует развитию жировой инфильтрации печени.
Таким образом, нарушения метаболизма аминокислот приводят к нарушению нормального функционирования организма человека.
Наследственные нарушения обмена аминокислот
(Ю.И. Барашнев, Ю.Е. Вельтищев, 1978 г.)
1. Наследственные нарушения обмена аминокислот, сопровождающиеся увеличением их концентрации в крови и моче: фенилкетонурия, гистидинемия, триптофанурия, болезнь "кленового сиропа", орнитинемия, цитруллинемия и др. Наследование, в основном, по аутосомно-рецессивному типу. В основе развития заболеваний лежит нарушение синтеза или структуры тех или иных ферментов.
2. Наследственные нарушения обмена аминокислот, сопровождающиеся увеличением их выделения с мочой без изменения уровня в крови: гомоцистинурия, гипофосфатазия, аргиносукцинатацидурия и др. При данных энзимопатиях нарушено обратное всасывание в почках, что приводит к увеличению их содержания в моче.
3. Наследственные нарушения систем транспорта аминокислот: цистинурия, триптофанурия, болезнь Гартнепа и др. К этой группе относятся энзимопатии, развитие которых обусловлено снижением реабсорбции аминокислот в почках и кишечнике.
4. Вторичные гипераминоцидурии: синдром Фанкони, фруктоземия, галактоземия, болезнь Вильсона-Коновалова и др. При данных состояниях возникает вторичная генерализованная гипераминоацидурия в результате вторичных тубулярных нарушений.
Фенилкетонурия (ФКУ)
Впервые описана в 1934 г. Folling под названием "фенилпировиноградная имбецильность". Тип наследования - аутосомнорецессивный. Частота заболевания составляет 1:10000- 1:20000 новорожденных. Пренатальный диагноз возможен при использовании генетических зондов и биопсии ворсин хориона.
К развитию классической клинической картины при ФКУ приводит недостаточность фенилаланингидроксилазы и недостаточность редуктазы дигидроптерина- 2-го фермента, обеспечивающего гидроксилирование фенилаланина. Их недостаток приводит к накоплению фенилаланина (ФА) в жидких средах организма (схема 1). Как известно, ФА относится к незаменимым аминокислотам. Поступающий с продуктами питания и не используемый для синтеза белка, он распадается по тирозиновому пути. При ФКУ наблюдается ограничение превращения ФА в тирозин и, соответственно, ускорение его превращения в фенилпировиноградную кислоту и другие кетоновые кислоты.
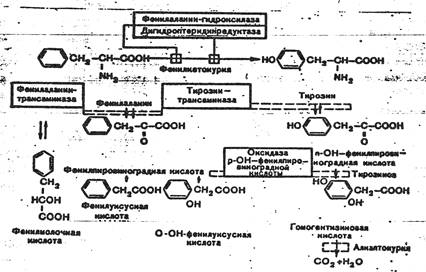
Схема 1. Варианты нарушений метаболизма фенилаланина.
Существование различных клинико-биохимических вариантов ФКУ объясняется тем, что фенилаланингидроксилаза является частью мультиферментной системы.
Различают следующие формы ФКУ:
1. Классическая
2. Скрытая.
3. Атипичная.
Развитие атипичных и скрытых форм ФКУ связывают с недостаточностью фенилаланинтрансаминазы, тирозинтрансаминазы и оксидазы парагидроксифенилпировиноградной кислоты. Атипичная ФКУ обычно не сопровождается поражением нервной системы в результате позднего развития ферментативного дефекта.
У женщин с фенилкетонурией возможно рождение детей с микроцефалией, задержкой умственного развития, нарушениями развития мочевыделительной системы, поэтому необходимо назначение диетотерапии во время беременности.
Клинические симптомы у больных ФКУ
При рождении ребенок с фенилкетонурией выглядит здоровым. Заболевание у этих детей проявляется на первом году жизни.
1. Интеллектуальный дефект. Нелеченный ребенок теряет около 50 баллов IQ к концу 1-го года жизни. У больных не выявляется зависимости между уровнем ФА и степенью интеллектуального дефекта.
2. Судорожный синдром (4 50%), экзема, гипопигментация.
3. Нарушение координации движения.
4. Задержка развития статических и двигательных функций.
5. Поражение пирамидных путей и стриопаллидарной системы. Клинические проявления классической ФКУ редко встречаются в странах, в которых действует программа неонатального скрининга на это заболевание.
У детей с фенилкетонурией наблюдается повышенный уровень в моче метаболитов ФА. Увеличение в физиологических жидкостях содержания ФА и недоокисленных продуктов его метаболизма приводит к поражению нервной системы. Определенная роль в этих нарушениях принадлежит дисбалансу аминокислот (дефицит тирозина, который в норме активно участвует в построении белкового компонента миелина). Демиелинизация является характерным патоморфологическим признаком фенилкетонурии. Нарушение соотношения аминокислот в
крови приводит к нарушению уровня свободных аминокислот в головном мозге, что вызывает слабоумие, гиперкинезы и другие неврологические симптомы.
Пирамидные симптомы обусловлены нарушением процессов миелинизации. Избирательный характер поражения нервной системы объясняется особенностями миелинизации- поражаются наиболее молодые в филогенетическом отношении отделы, выполняющие сложные и дифференцированные функции. С недостаточным образованием меланина из тирозина связывают голубой цвет глаз, светлую кожу. Запах "плесени" ("мышиный", "волчий") объясняется наличием фенилуксусной кислоты в моче. Кожные проявления (экссудативный диатез, экземы) связаны с выделением аномальных метаболитов. Недостаточность образования адренергических гормонов из тирозина приводит к артериальной гипотонии.
Необходимо отметить, что при ФКУ в патологический процесс вовлекается печень, но характер морфологических расстройств не является специфичным: выявляются признаки тканевой гипоксии, нарушения окислительной и белоксинтезирующей функции, перегрузка липидами. Наряду с этим наблюдаются компенсаторно-приспособительные изменения: высокое содержание гликогена, гиперплазия митохондрий. Генерализованную гипераминоацидемию при ФКУ можно объяснить вторичным нарушением метаболизма аминокислот в связи с повреждением гепатоцитов, т.к. многие ферменты, участвующие в аминокислотном обмене, локализуются в печени.
У нелеченых больных с классической ФКУ наблюдается значительное снижение концентрации катехоламинов, серотонина и их производных в моче, крови, ликворе. Поэтому в комплексном лечении ФКУ необходима промедиаторная коррекция, так как парциальный интеллектуальный дефект может быть связан с нейромедиаторными нарушениями.
Критерии диагностики классической формы фенилкетонурии:
1. Уровень ФА в плазме выше 240 ммоль/л.
2. Вторичный дефицит тирозина.
3. Повышенный уровень в моче метаболитов ФА.
4. Сниженная толерантность к полученному внутрь ФА.
Методы диагностики фенилкетонурии:
1. Проба Феллинга с FeCl3 - при положительном анализе появляется сине-зеленое окрашивание мочи.
2. В крови выявление избытка фенилаланина возможно с помощью бактериального экспресс- теста Гольдфарба или теста Гатри (т.к. в течение первых дней жизни фенилпировиноградная кислота в моче может отсутствовать).
При ФКУ проводится лечение диетой с ограниченным содержанием ФА (главным образом назначают овощные блюда, мед, фрукты). Такие продукты, как молоко, молочные изделия, яйца, рыба, должны быть полностью исключены в период пребывания больных с ФКУ на острой диете. Назначаются специальные препараты (цимогран, лофеналак) и витамины.
Оптимальные сроки обследования новорожденных- 6-14 день жизни, начало терапии - не позднее 21 дня жизни. Необходимо помнить, что проведение исследования в первые сутки не исключает ложноположительных или ложноотрицательных результатов ( повторное исследование проводят до 21 дня жизни ). Эффективность лечения оценивается по интеллектуальному уровню развития пациента. Необходимо отметить, что лечение, начатое после года не нормализует интеллект полностью (возможно, это связано с развитием необратимых изменений в мозге).
ГОМОЦИСТИНУРИЯ
Заболевание впервые описано в 1962 г. Carson и Neil. Наследуется по аутосомно-рецессивному типу. Частота гомоцистинурии составляет 1:200 000 новорожденных.
В основе заболевания лежит отсутствие или снижение активности фермента цистатионинсинтетазы, что ведет к нарушению обмена метионина. Кофактором цистатионсинтетазы является витамин В6 . Поэтому наблюдается пиридоксинчувствительная и пиридоксинрезистентная формы. У родителей и родственников больных часто обнаруживают шизофрению. Рядом авторов отмечается фенотипическое сходство с болезнью Марфана. Однако, при гомоцистинурии в отличие от болезни Марфана более выражены изменения нервной системы, снижение интеллекта и судорожный синдром.
Формы гомоцистинурии:
1. Классическая.
2. Связанная с дефицитом утилизации витамина В6 .
3. Обусловленная нарушением метаболизма фолиевой кислоты.
Клинически дети, больные классической формой, при рождении выглядят здоровыми. Возможны лишь задержка роста и развития. Диагноз обычно устанавливается после 3-х лет, когда выявляется подвывих хрусталика. Основной рентгенологический признак- генерализованный остеопороз. При гомоцистинурии наблюдается клинический полиморфизм. Однако, можно выделить своеобразный комплекс признаков (таблица 1).
Таблица 1. Симптомокомплекс гомоцистинурии
( Ю.И.Барашнев, Ю.И.Вельтищев, 1978 г.)
| Симптомы | Фенотипические проявления |
| Изменение скелета | Укороченное туловище,удлиненные конечности, нарушенная осанка, "башенная" форма черепа, неправильный прикус и рост зубов,высокое небо, "крыловидные" лопатки, воронкообразная деформация грудной клетки, вальгусная деформация грудной клетки, вальгусная деформация нижних конечностей, плоскостопие, умеренный остеопороз |
| Изменение нервной системы | Сниженный интеллект,патологический характер ЭЭГ, спастическая походка |
| Нарушение зрения | Подвывих хрусталиков, вторичная глаукома, изменения глазного дна |
| Сердечно- сосудистые расстройства | Нарушение обменных процессов в миокарде (по данным ЭКГ) |
| Внешние признаки | Светлые, мягкие,вьющиеся крупными завитками волосы, голубой цвет радужной оболочки |
Гомоцистинурия ведет к поражению соединительной ткани за счет усиления синтеза сульфатированных протеогликанов с последующей дегенерацией эластических элементов, депозицией коллагена и кальцификацией. Вовлечение в патологический процесс соединительной ткани приводит к костным деформациям, подвывиху хрусталика книзу. Генез подвывиха хрусталика следующий: дефицит цистина приводит к вовлечению в процесс соединительнотканных волокон цинновых связок. У больных с гомоцистинурией выявляются разнообразные поражения органа зрения: близорукость, изменения глазного дна, вторичная глаукома. Резкое падение остроты зрения приводит к инвализации больных.
При гомоцистинурии с мочой избыточно выводится гомоцистин в результате инактивации цистатионинсинтетазы в головном мозге и печени больных. Изменения ЦНС у детей с гомоцистинурией обнаруживаются в 50 % случаев. Происхождение интеллектуального дефекта при этом заболевании до сих пор не ясно. Отмечено сочетание гомоцистинурии с эпилепсией, шизофренией, задержкой интеллектуального развития. Высказываются предположения, что основной причиной умственного дефекта при гомоцистинурии являются сосудистые мозговые тромбы, в связи с чем образуется много мелких инфарктов мозга. При этом возникают некротические и дегенеративные изменения в мозговом стволе, зрительном бугре и коре.
У больных более старшего возраста (до 30 лет) наблюдается склонность к артериальным и венозным тромбозам, что связано с активацией гомоцистином фактора Хагемана, а также с оседанием гомоцистина из-за его низкой растворимости в патологически измененной интиме сосудов. Оба этих процесса могут создавать условия для образования тромбов.
Необходимо отметить, что гомоцистинурии свойственен прогредиентный характер течения патологического процесса. При рождении дети обычно не имеют каких-либо внешних дефектов. Но в результате наследственных нарушений постепенно начинают выявляться изменения в отдельных органах и системах. Существует зависимость клинической картины от возраста. Чем старше ребенок, тем больше органов и систем вовлекается в патологический процесс. Таким образом, гомоцистинурия - это заболевание всего организма. Диагностическим признаком является избыточная экскреция с мочей гомоцистина (необходима свежесобранная моча, т.к. гомоцистин разрушается при хранении).
ГИСТИДИНЕМИЯ
Заболевание впервые описано в 1961 г. Ghadimi с соавторами, а термин "гистидинемия" предложен Auerbach с соавт. в 1962 году.
Заболевание возникает в результате отсутствия или недостаточности активности фермента гистидазы. Наследуется аутосомно-рецессивно. Высказывается мнение об аутосомно-рецессивной передаче с неполной пенетрантностью, сцепленном с Х-хромосомой наследовании. Т.е., возможно, различные формы гистидинемии наследуются по разному. Для детей первого года жизни гистидин - незаменимая аминокислота. При недостатке гистидина в этом возрасте отмечается нарушение ретенции азота, выявляется дефицит массы тела, появляется шелушение кожи и экзематозные высыпания. Существует точка зрения о незаменимости гистидина и для здоровых взрослых (при недостатке наблюдаются признаки анемии с нарушением эритропоэза).
При гистидинемии происходит нарушение самого эффективного активного пути катаболизма- превращение гистидина в уроканиновую кислоту (этим путем в норме катаболизируется большая часть гистидина). В результате метаболического блока происходит накопление в крови и моче гистидина. Увеличение активности трансаминирования и усиленный перевод гистидина в имидазолпировиноградную, имидозолмолочную и имидозолуксусную кислоты является компенсаторно-приспособительной реакцией организма.
Гистидинемия отличается большой вариабельностью клинических проявлений: от тяжелой умственной отсталости до полного отсутствия каких-либо симптомов. Снижение интеллекта выявляется лишь у 50 % больных детей. Больные гистидинемией имеют светлый цвет волос, голубые глаза. На первый план у таких детей выступает поражение нервной системы: снижение интеллекта, нарушение речи, судороги. А у детей с нормальным интеллектом можно выявить особенности психики при гистидинемии: эмоциональную лабильность, агрессивность. Характер повреждения нервной системы при данном заболевании зависит от степени инактивации гистидидазы. Иногда гистидинемия сочетается с аномалиями развития, патологией почек, костной системы.
Для диагностики заболевания необходимо выявление повышенного уровня гистидина в плазме. Окончательный диагноз подтверждает определение гистидидазы в ороговевающем эпителии или печени.
Наследственные нарушения обмена триптофана
Как известно, триптофан является незаменимой аминокислотой.
Образующийся при расщеплении белков триптофан через кишечную стенку всасывается в кровь и используется организмом для синтеза белков. Остальная часть идет на синтез биологически активных веществ: никотинамида, серотонина, мелатонина и др. Таким образом, процессы метаболизма триптофана идут по трем основным направлениям: серотониновому, индольному и кинурениновому.
К наследственно обусловленным нарушениям обмена триптофана относится ряд клинических синдромов и заболеваний:
1. Болезнь Гартнепа.
2. Индиканурия.
3. Синдром Тада.
4. Синдром Прайса.
Болезнь Гартнепа
Впервые описана Baron с соавт. в 1956 году. Аутосомно- рецессивный тип наследования. При данном заболевании наблюдается генетическое изменение транспортной функции клеток слизистой оболочки кишечника и проксимальных отделов почечных канальцев. Это ведет к изолированному дефекту транспорта моноаминокарбоновых кислот. Нарушение кишечной абсорбции триптофана приводит к его бактериальному расщеплению в кишечнике до индола и индоксила. Генерализованная аминоацидурия обусловлена нарушением канальцевой реабсорбции.
Для болезни Гартнепа характерны кожная фоточуствительность, пеллагроподобный дерматит, мозжечковая атаксия с вовлечением в процесс пирамидных путей, нарушение функции желудочно-кишечного тракта. У некоторых детей выявляется умственная отсталость.
Индиканурия
Впервые описана в 1965 году Bickel.
В основе заболевания лежит нарушение всасывания триптофана в кишечнике с образованием избыточного количества индола, который всасывается, окисляется, сульфатируется и выделяется в виде индикана. Последний окисляется под влиянием воздуха до голубого индикана, окрашивающего пеленки в синий цвет (болезнь "голубых пеленок").
При индиканурии наблюдается гиперкальциемия, нефрокальциноз, периодическая гипертермия.
Синдром Тада
Данный синдром впервые описан в 1963 году Tada с соавт. под названием "триптофанурия с нанизмом". Аутосомно- рецессивный тип наследования. При синдроме Тада наблюдается недостаток фермента триптофанпирролазы, катализирующего превращение триптофана в кинуренин. Нарушения связаны с эндогенным дефицитом никотиновой кислоты и избытком индольных соединений. При синдроме Тада отмечается глубокая умственная отсталость, нанизм, мозжечковая атаксия.
Синдром Прайса
Впервые описан в 1967 г. Price с соавторами. Генетический дефект кинуренингидроксилазы. Наблюдается избыточное выделение с мочой кинуренина за счет блока фермента. Основное проявление синдрома Прайса - склеродермия.
Галактоземия
В 1908 году Reuss впервые отметил, что галактоземия относится к "врожденным ошибкам обмена". Изучению основного и побочного путей преобразования галактозы способствовали труды многих ученых.
Классическая галактоземия наследуется по аутосомно- рецессивному типу. По частоте занимает 2-е место после ФКУ. При этом заболевании наблюдается дефицит галактозо-1-фосфат-уридилтрансферазы. В эритроцитах гомозигот при классической галактоземии активность данного фермента почти не определяется, у гетерозигот составляет 50 % нормы.
Главный путь преобразования галактозы в глюкозу - это путь Лелуа. Первым этапом является фосфорилирование галактозы в печени, мозге, эритроцитах. В результате этой реакции образуется галактозо-1-фосфат, для дальнейшего превращения которого необходим специфический фермент галактозо-1-фосфат- уридилтрансфераза. При дефиците галактозо-1-фосфат - уридилтрансферазы невозможен дальнейший метаболизм галактозо-1-фосфата до УДФ- галактозы. Накопление галактозо-1-фосфата приводит к заболеванию, возможно, и во внутриутробном периоде. Кроме основного пути обмена галактозы (пути Лелуа), существуют обходные, дополнительные пути, которые необходимо учитывать в патологических условиях при дефиците галактозы.
Необходимо помнить, что из-за наличия большого числа фенокопий галактоземия не всегда распознается своевременно. Целый ряд болезней периода новорожденности клинически сходен с галактоземией: гемолитическая болезнь новорожденных, врожденный гепатит, атрезия желчных путей, энзимопатическая желтуха Криглера-Найяра и ряд других заболеваний.
При классической галактоземии наблюдается раннее появление симптомов заболевания: анорексия, рвота, непереносимость молока, задержка увеличения массы тела, катаракта, гепатомегалия, желтуха, отеки, геморрагические проявления, спленомегалия, поражение ЦНС. Задержка психомоторного развития выявляется рано и прогрессирует с возрастом (однако, умственная отсталость менее выражена, чем при ФКУ).
Тяжелые формы заболевания имеют яркую клиническую картину уже в первые дни жизни: расстройство пищеварения и резкая интоксикация (рвота, понос, гипотрофия), желтуха, которая имеет стойкий характер и наступает за счет увеличения концентрации прямого билирубина. Нередко печеночная желтуха сочетается с гемолитической за счет снижения осмотической резистентности эритроцитов. Необходимо отметить, что галактозо-1-фосфат является гепатотоксичным. Постепенно у больного прогрессируют признаки печеночной недостаточности. На 2-3 месяце жизни развивается асцит, спленомегалия, нарастают явления токсикоза и эксикоза. Повышенный уровень галактиола приводит к развитию катаракты (при тяжелой форме галактоземии катаракта может наблюдаться при рождении). Терминальная стадия болезни характеризуется печеночной недостаточностью, выраженной дистрофией и присоединением вторичных инфекций.
Диагностика заболевания основана на определении галактозы в крови и моче. Пренатальная
8-09-2015, 22:28