Йодхлорометрия — отличается от йодометрии использованием в качестве титранта не раствора йода, а более устойчивого раствора йодмонохлорида. Аналогично йоду йодмонохлорид образует йодпроизводные органических оснований. Избыток титранта устанавливают йодометрически:
![]()
Йодатометрия основана на окислении органических соединений йодатом калия. Избыток титранта устанавливают йодометрически:
![]()
Брочаточетрия (бромид-броматометрия) основана на использовании окислительных свойств или реакции замещения -р.-^гкиг моно-.ди- или трибромпроизводных) за счет образующегося свободного брома:
![]()
![]()
Индикаторами при прямом титровании служат азокрасители, которые обесцвечиваются бромом в эквивалентной точ- • е метиловый красный). В случае обратного титрования эквивалентную точку устанавливают йодометрически по избыт- ?г. -'.гранта (бромата калия).
Перманганатометрия основана на использовании окислительных свойств титранта — перманганата калия в кислой среде:
Индикатором при прямом титровании служит сам титрант (появляется фиолетовое окрашивание), а при обратном титровании избыток титранта устанавливают йодометрическим методом.
Цериметрия основана на использовании окислительных свойств титранта — соли церия (IV), который в кислой среде восстанавливается до церия (III):
![]()
Индикатором служат дифениламин, о-фенантролин, а при обратном титровании избыток титранта устанавливают йодометрически:
![]()
22 Комплексономе трия
Метод основан на образовании прочных, растворимых в воде комплексов катионов металлов с трилоном Б — динатри- евой солью этилендиаминтетрауксусной кислоты (ЭДТАЫаг) или другими комплексонами. Независимо от заряда катиона взаимодействие его с титрантом (ЭДТА №2) происходит в стехиометрическом соотношении 1:1:
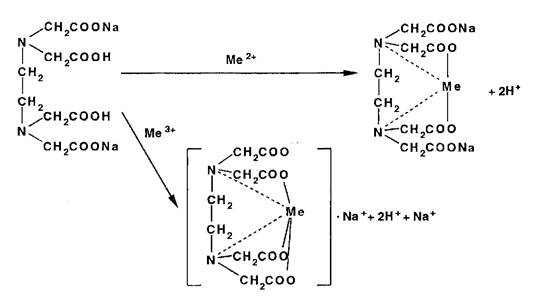
Используют метод для количественного определения неорганических и органических ЛВ, содержащих катионы магния, кальция, цинка, висмута, свинца, алюминия и др. Точку эквивалентности устанавливают с помощью металлоинди- каторов — органических красителей (ксиленоловый оранжевый, пирокатехиновый фиолетовый, кислотный хром темно-синий). образующих с указанными катионами непрочные, ярко окрашенные комплексы. В эквивалентной точке эти комплексы разрушаются до образования свободного индикатора, по окраске которого делают заключение о конце титрования.
Непременным условием комплексонометрии является строгое соблюдение при титровании определенного интервала рН, что достигается с помощью буферных растворов. Комплексонометрическое титрование может быть выполнено прямым, обратным и косвенным (заместительным) методами.
23 Нитритометрия
Метод количественного определения первичных и вторичных ароматических аминов, основанный на использовании титранта — раствора нитрита натрия в присутствии бромида натрия:
![]()
![]()
![]()
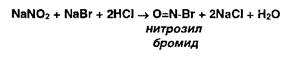
В этих условиях с первичными ароматическими аминами образуются диазосоединения (в кислой среде):
Вторичные ароматические амины в тех же условиях образуют Ы-нитрозосоединения:
Эквивалентную точку устанавливают различными путями: потенциометрически, с помощью указанных в ФС внутренних индикаторов (тропеолин 00, нейтральный красный), с внешним индикатором (йодкрахмальная бумага). Титрование с йодкрахмальной бумагой ведут до тех пор, пока капля титруемого раствора, взятая через 1 мин после прибавления титранта, не вызовет тотчас же посинение бумаги:
На результаты определения влияют температура (смесь охлаждают до 5-15°С), концентрация хлороводородной кислоты, природа растворителя.
При использовании внутренних индикаторов наблюдают изменение их окраски в эквивалентной точке.
24 Элементный анализ
Определение азота в органических соединениях (метод Кьельдаля)
Метод основан на предварительной минерализации азотсодержащего органического соединения до гидросульфата аммония. Определение выполняют с помощью прибора, состоящего из колбы Кьельдаля, парообразователя, холодильника, приемника. Оно состоит из нескольких стадий.
1. Минерализация (нагревание с конц. Н2504):
![]()
2. Разложение (1ЧН4)Н304 гидроксидом натрия и отгонка образующегося аммиака в приемник:
![]()
3. Взаимодействие ЫНз в приемнике с борной кислотой с образованием тетрагидроксибората аммония:
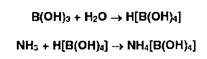
4. Титрование отгона ОДМ раствором хлороводородной кислоты:
![]()
Параллельно выполняют контрольный опыт (без анализируемого вещества) для повышения точности результатов анализа.
Для определения веществ, содержащих в молекуле амидную группу, используют упрощенный вариант метода Кьельдаля, исключающий стадию минерализации. Методика сводится к гидролизу амида в колбе Кьельдаля 30% раствором гид- роксида натрия, отгонке выделяющегося аммиака или амина в приемник и титровании отгона 0,1 М хлороводородной кислотой.
25 Метод сжигания в колбе с кислородом
Используется для анализа ЛВ, содержащих в молекуле галогены, серу, фосфор. Сжигание проводят в колбе из термо- ^ никого стекла, наполненной кислородом. В пробку колбы впаяна платиновая или нихромовая проволока, заканчиваются спиралью (держатель), в которую помещают точную навеску ЛВ, завернутую в фильтровальную бумагу. На дно колбы вливают поглощающую жидкость. По окончании сжигания колбу оставляют на 30-60 мин, периодически перемеши- г:- После этого химическим или физико-химическим методом идентифицируют или определяют образовавшиеся ионы.
Так. например, йодсодержащие органические соединения последовательно количественно превращают в йодаты.
Сжигание ЛВ в атмосфере кислорода приводит к окислению до свободного йода, растворяющегося в растворе гид- :: ксида натрия (поглощающая жидкость) с образованием йодида и гипойодита натрия:
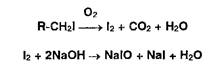
2. Для окисления образовавшихся йодидов до йодатов в колбу вносят раствор ацетата брома до появления желтого окрашивания:
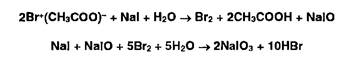
3. Для удаления избытка брома добавляют концентрированную муравьиную кислоту до обесцвечивания раствора:
![]()
4. Выдерживают 5 мин в темном месте после добавления йодида калия и раствора серной кислоты, а затем титруют выделившийся йод, содержание которого эквивалентно его количеству в испытуемом ЛВ:
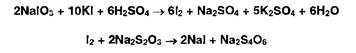
26 Физические и физико-химические методы анализа
Физические и физико-химические методы могут быть классифицированы на следующие группы: оптические методы, методы, основанные на поглощении электромагнитного излучения, методы, основанные на испускании излучения, методы, основанные на использовании магнитного поля, электрохимические методы, термические методы, методы разделения.
Физико-химические методы основаны на использовании зависимости физических свойств от химического состава веществ. В большинстве случаев физико-химические методы отличаются быстротой выполнения, избирательностью, высокой чувствительностью, возможностью унификации и автоматизации. Поэтому данная группа методов приобретает все большее значение для объективной оценки качества ЛС, в т.ч. для испытания на подлинность, испытания на чистоту и для количественного определения.
27 Оптические методы
Рефрактометрия основана на наличии зависимости величины показателя преломления света от концентрации раствора испытуемого вещества. Показатель преломления зависит также от температуры, длины волны света, концентрации вещества и природы растворителя. Рефрактометрию используют для установления подлинности лекарственных веществ по молярной рефракции. Для количественного определения выбирают интервал линейной зависимости между концентрацией раствора и коэффициентом преломления. В этом интервале концентрацию (х) вычисляют по формуле: х=(п — по)/Р, где л — показатель преломления раствора вещества; по — показатель преломления растворителя; Б — фактор, равный величине прироста показателя преломления при увеличении концентрации вещества на 1% (устанавливается экспериментально).
Рефрактометрические определения выполняют на рефрактометрах, при стабильной температуре (20±0,3°С) и длине ? злны линии Б спектра натрия (589,3 нм) в диапазоне показателей преломления от 1,3 до 1,7. Прибор юстируют по эта- - м~:ным жидкостям или воде очищенной, для которой ПО20 = 1,3330.
Поляриметрия — метод, основанный на способности вещества вращать плоскость поляризованного света. Эта способ- -: ;гь обусловлена наличием в молекулах ассиметрических атомов углерода. Степень отклонения плоскости поляризации
первоначального положения выражается в угловых градусах. Эту величину называют углом вращения (а). Правовращающие вещества вращают плоскость поляризации по часовой стрелке (обозначают знаком +), левовращающие — против часовой стрелки (-).
Для растворов величина а зависит от природы растворителя, концентрации оптически активного вещества и длины рабочего слоя кюветы с раствором. Подлинность и чистоту лекарственных веществ подтверждают по величине удельного вращения [а]о20 , измеренного при 20"С и длине волны Б спектра натрия. Величину [а] о20 для растворов веществ рассчитывают по формуле:
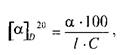
где а — измеренный угол вращения, в градусах; / — длина рабочего слоя кюветы, в дециметрах; С — концентрация раствора вещества (г/100 мл).
Количественно определяют (в %) содержание оптически активного вещества в растворе по формуле:
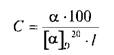
Величину а измеряют на поляриметрах с точностью до ±0,02°.
28 Методы, основанные на поглощении электромагнитного излучения
Используют спектрофотометрические методы анализа по поглощению веществами монохроматического электромагнитного излучения (в УФ- и ИК-области) и фотоколориметрические (колориметрические) методы анализа по поглощению веществами немонохроматического излучения.
Фотометрические методы анализа основаны на использовании объединенного закона Бугера-Ламберта-Бера:
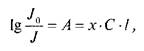
где/о — интенсивность излучения, падающего на вещество; /— интенсивность излучения, прошедшего через вещество; А — величина оптической плотности; х — показатель поглощения данного вещества; С — концентрация раствора анализируемого вещества, г; I — длина рабочего слоя кюветы, см.
На основании этого закона содержание вещества в растворе определяют по формуле:
![]()
В случае несоответствия закону Бугера-Ламберта-Бера вначале с помощью стандартного раствора устанавливают зависимость оптической плотности от концентрации, а затем строят калибровочный график, с помощью которого выполняют расчеты.
Спектрофотометрия в УФ- и видимой областях — один из наиболее широко используемых физико-химических методов в фармацевтическом анализе.
Анализируемые ЛВ должны иметь в структуре молекулы хромофорные группы (сопряженные связи, ароматическое ядро и др.), обусловливающие различные электронные переходы в молекулах и поглощение электромагнитного излучения.
Кривая зависимости интенсивности светопоглощения от длины волны (нм) называется спектром поглощения вещества и является его специфической характеристикой. Измерение спектров поглощения растворов анализируемых веществ в ультрафиолетовой (190-380 нм) и видимой (380-780 нм) областях производят с помощью спектрофотометров различных марок (СФ-26, СФ-46 и др.). В качестве растворителей используют свободные от примесей воду, растворы кислот и щелочей, этанол, хлороформ и другие органические растворители.
Спектрофотометрической константой является удельный показатель поглощения ( ), который рассчитывают по формуле:
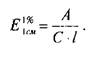
Удельный показатель поглощения представляет собой величину оптической плотности раствора, содержащего
1,0 г вещества в 100 мл раствора, измеренную в кювете с рабочей длиной 1 см. Установив по стандартному образцу величину и преобразовав эту формулу, можно рассчитать концентрацию анализируемого вещества с относительной погрешностью до ±2%.
Идентификацию ЛВ можно провести по Д1 ^, характеру спектральных кривых в различных растворителях, положению максимума и минимума светопоглощения или их отношению (при различных длинах волн). Для количественного спек- трофотометрического анализа важен выбор аналитической полосы поглощения. Последняя должна быть свободна от наложения полос поглощения других компонентов смеси и иметь достаточно высокий удельный показатель поглощения анализируемого вещества.
Фотоколориметрия отличается от спектрофотометрического анализа тем, что анализируемое вещество с помощью какого-либо реагента переводят (количественно) в окрашенное соединение. Вначале получают окрашенные растворы, используя растворы стандартных образцов (ГСО или PCO). Измерение оптической плотности производят на фотоколориметрах. Затем строят калибровочный график зависимости интенсивности поглощения окрашенных растворов от концентрации, по которому рассчитывают содержание JTB в испытуемых образцахJIBили ЛФ.
Метод дифференциальной спектрофотометрии и фотоколориметрии основан на измерении светопоглощения анализируемого раствора относительно раствора сравнения, содержащего определенное количество стандартного образца испытуемого вещества или его заменителя. Такой прием приводит к изменению рабочей области шкалы прибора и снижению относительной погрешности определения до ±0,5-1%, т.е. сопоставимой с титриметрическими методами.
Производная УФ-спектрофотометрия является одним из вариантов дифференциальной спектрофотометрии. Если в дифференциальной спектрофотометрии используют разность оптических плотностей при одной и той же длине волны, то в производной — при двух длинах волн, разделенных небольшим интервалом. Этот вариант основан на выделении индивидуальных полос из УФ-спектра, который представляет собой сумму налагающихся полос поглощения или полос, не имеющих четко выраженного максимума. При этом на спектральных кривых в координатах производная-длина волны появляются полосы с отчетливо выраженными максимумами и минимумами. Благодаря этому можно идентифицировать сходные по химической структуре вещества, повысить избирательность анализа и выполнять количественное определение двух-, трехкомпонентных смесей более экономично и эффективно, чем титриметрическими методами.
Одним из вариантов дифференциальной спектрофотометрии является АЕ-метод. Он основан на превращении одного из веществ, входящих в состав анализируемой пробы, в таутомерную (или иную) форму, отличающуюся по характеру и интенсивности светопоглощения. Затем измеряют светопоглощение раствора одной таутомерной формы по отношению к другой, т.е. используют в качестве стандарта раствор анализируемого вещества.
Спектрофотометрии в ИК-области. Природа полос поглощения в ИК области связана с колебательными переходами и изменением колебательных состояний ядер, входящих в молекулу поглощающего вещества. Поэтому поглощением в ИК-области обладают молекулы, дипольные моменты которых изменяются при возбуждении колебательных движений ядер. Область применения ИК-спектроскопии аналогична, но более широка, чем у УФ-метода. ПК-спектр однозначно характеризует всю структуру молекулы, включая незначительные ее изменения. Важные преимущества ИК-спектроскопии - - высокая специфичность, объективность полученных результатов, возможность анализа веществ в кристаллическом состоянии. Для измерения ИК-спектров на однолучевых или двулучевых ИК-спектрофотометрах используют взвеси веществ в вазелиновом масле или помещают анализируемое вещество между пластинами из бромида калия. Каждый ИК-спектр представляет собой серию полос поглощения, максимумы которых определяются волновым числом, измеряемым в см"1 , и определенной интенсивностью. Для анализа ЛВ обычно используют спектральную область от 4000 до 400 см-1 .
ГФ XI рекомендует два способа установления подлинности по ИК-спектрам. Один из них основан на сравнении зарегистрированных в идентичных условиях ИК-спектров испытуемого ЛВ и его стандартного образца. Второй способ заключается в сравнении ИК-спекгра испытуемого ЛВ с его стандартным спектром, прилагаемым к ФС и зарегистрированным в соответствии с указанными в ней требованиями.
Фототурбидиметрия — метод, основанный на измерении интенсивности света, поглощенного тонкодисперсной суспензией, и фотонефелометрия — метод, основанный на измерении света, рассеянного взвешенными частицами анализируемого вещества. Оба метода применяют в фармацевтическом анализе для количественного определения ЛВ, образующих с различными реактивами тонкие суспензии. Предварительно устанавливают зависимость между интенсивностью поглощения (рассеяния) света и концентрацией вещества в анализируемом растворе. Способы расчета аналогичны фотометрическим методам.
30 Методы, основанные на испускании излучения
Атомно-абсорбционная спектрометрия основана на поглощении атомами излучения с частотой, равной частоте резонансного перехода. Излучение исходит от лампы с полым катодом, проходит через пламя, в котором распыляется проба, пропускается через щель монохроматора, и выделенная из спектра резонансная линия определяемого элемента измеряется фотоэлектрическим способом. Затем устанавливается зависимость между ослаблением интенсивности излучения источника света и концентрацией испытуемого вещества.
Флуоресцентные методы основаны на способности веществ флуоресцировать в УФ-свете, обусловленной либо химической структурой самих органических веществ, либо продуктов их диссоциации, сольволиза, других превращений. Способностью флуоресцировать обладают обычно органические соединения с симметричной структурой молекул, в которых имеются сопряженные связи (нитро-, нитрозо-, азо-, амидные, карбонильные или карбоксильные группы).
Флуориметрия используется не только для установления подлинности, но и определения малых количеств веществ, т.к. интенсивность флуоресценции имеет линейную зависимость от концентрации. Линейная зависимость сохраняется при постоянстве квантового выхода и интенсивности возбуждающего света для низких концентраций веществ. При высоких концентрациях эта зависимость нарушается. Идентификацию проводят по цвету излучаемого света, специфичного для флуоресцирующих веществ. Спектр дает широкие полосы излучения (от 100 до 200 нм). Метод отличается очень высокой чувствительностью. Количественное определение выполняют на спектрофлуориметрах. Расчет концентрации производят с помощью калибровочного графика или шкалы стандартных растворов, аналогично фотометрическим методам.
31 Методы, основанные на использовании магнитного поля
Спектроскопия ядерного магнитного резонанса (ЯМР) — метод, основанный на регистрации индуцированных радиочастотным полем переходов между ядерными магнитными энергетическими уровнями молекул вещества, помещенного в магнитное поле. Метод позволяет изучать магнитные переходы ядер со спиновыми квантовыми числами больше нуля (ядра 'Н,13 С,19 Р, 31 Р). Совокупность сигналов переходов между энергетическими уровнями ядер молекул составляет спектр ЯМР. Каждый спектр ЯМР регистрируется для одного типа ядер и специфичен для каждого вещества. Чаще всего используют спектроскопию на протонах (ПМР) и ЯМР 13 С.
Спектры регистрируют при помощи ЯМР-спектрометров. Каждый спектр является отражением числа ядер, порядка их связи и геометрии расположения ядер в молекуле. Спектр представляет собой совокупность пиков с различной шириной, площадью и интенсивностью сигналов. По характеру протонных сигналов можно сделать заключение о наличии в молекуле тех или иных групп атомов. Величина химического сдвига имеет порядок 10~6 или млн~! (миллионная доля). Она зависит от наличия в молекуле тех или иных групп, например, химический сдвиг у ароматических протонов находится в интервале 7-9 млн4 , альдегидных — 9-10 млн-1 и т.д.
Метод ЯМР- и ПМР-спектроскопии используют для объективной идентификации органических Л В и для количественного определения относительного содержания вещества или примеси. Подлинность может быть подтверждена либо пут ем сравнения со стандартным образцом, либо по наиболее характерным сигналам спектра, либо по полному набору спектральных параметров.
Масс-спектроскопия — метод, позволяющий определить массу ионов, ионизированных молекул или фрагментов молекул по отклонению в магнитных и электрических полях или по кинетической энергии. Ионизация молекул происходит в результате воздействия пучка электронов. Интенсивность пика в масс-спектре пропорциональна ч ислу образовавшихся ионов данного вида. Состав и массовые числа характеристических ионов позволяют установить принадлежность исследуемого соединения к определенному классу веществ, осуществить его идентификацию. Масс-спектроскопия отличается большой информативностью и очень высокой чувствительностью.
32 Электрохимические методы
Потенциометрия — метод, основанный на измерении равновесных потенциалов, возникающих на границе между испытуемым раствором и погруженным в него электродом. В фармацевтическом анализе наиболее широко используют по- тенциометрическое титрование. Оно основано на установлении эквивалентного объема титранта путем измерения ЭДС, возникающей при титровании за счет разности потенциалов индикаторного электрода и электрода сравнения, погруженных в анализируемый раствор. Метод
8-09-2015, 23:04