Под влиянием соматостатина секреция инсулина понижается. β-клетки также находятся под влиянием автономной нервной системы. Парасимпатическая часть (холинергические окончания блуждающего нерва) стимулирует выделение инсулина. Симпатическая часть (адреналин через α2 -адренорецепторы) подавляет выделение инсулина.
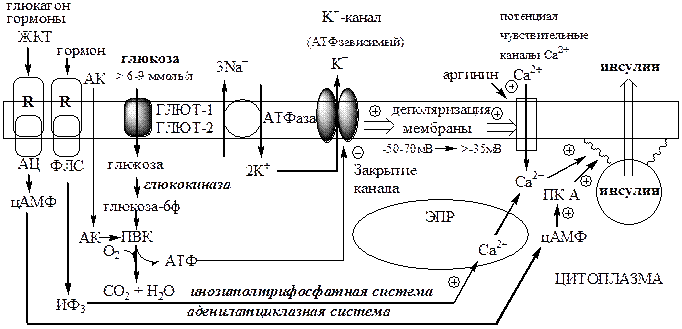
Секреция инсулина осуществляется с участием нескольких систем, в которых основная роль принадлежит Са2+ и цАМФ.
Поступление Са2+ в цитоплазму контролируется несколькими механизмами:
1). При повышении концентрации глюкозы в крови выше 6-9 ммоль/л, она при участии ГЛЮТ-1 и ГЛЮТ-2 поступает в β-клетки и фосфорилируется глюкокиназой. При этом концентрация глюкозо-6ф в клетке прямо пропорциональна концентрации глюкозы в крови. Глюкозо-6ф окисляется с образованием АТФ. АТФ образуется также при окислении аминокислот и жирных кислот. Чем больше в β-клетке глюкозы, аминокислот, жирных кислот тем больше из них образуется АТФ. АТФ ингибирует на мембране АТФ-зависимые калиевые каналы, калий накапливается в цитоплазме и вызывает деполяризацию клеточной мембраны, что стимулирует открытие потенциалзависимых Са2+ -каналов и поступление Са2+ в цитоплазму.
2). Гормоны, активирующие инозитолтрифосфатную систему (ТТГ), выпускают Са2+ из митохондрий и ЭПР.
цАМФ образуется из АТФ с участием АЦ, которая активируется гормонами ЖКТ, ТТГ, АКТГ, глюкагоном и Са2+ -кальмодулиновым комплексом.
цАМФ и Са2+ стимулируют полимеризацию субъединиц в микротубулы (микроканальцы). Влияние цАМФ на микроканальцевую систему опосредуется через фосфорилирование ПК А микроканальцевых белков. Микроканальцы способны сокращаться и расслабляться, перемещая гранулы по направлению к плазматической мембране обеспечивая экзоцитоз.
Секреция инсулина в ответ на стимуляцию глюкозой представляет собой двухфазную реакцию, состоящую из стадии быстрого, раннего высвобождения инсулина, называемую первой фазой секреции (начинается через 1 мин, продолжается 5-10 мин), и второй фазы (продолжительность ее до 25-30 мин).
Транспорт инсулина. Инсулин водорастворим и не имеет белка-переносчика в плазме. Т1/2 инсулина в плазме крови составляет 3—10 мин, С-пептида — около 30 мин, проинсулина 20-23 мин.
Разрушение инсулина происходит под действием инсулинзависимой протеиназы и глутатион-инсулин-трансгидрогеназы в тканях мишенях: в основном в печени (за 1 проход через печень разрушается около 50% инсулина), в меньшей степени в почках и плаценте.
БИОЛОГИЧЕСКИЕ ФУНКЦИИ ИНСУЛИНА
Инсулин — главный анаболический гормон, он влияет на все виды обмена веществ во всём организме. Однако в первую очередь действие инсулина касается обмена углеводов.
Влияние инсулина на метаболизм глюкозы
Инсулин стимулирует утилизацию глюкозы в клетках разными путями. Около 50% глюкозы используется в процессе гликолиза, 30—40% превращается в жиры и около 10% накапливается в форме гликогена. Общий результат стимуляции этих процессов — снижение концентрации глюкозы в крови.
Влияние инсулина на метаболизм липидов
В печени и жировой ткани инсулин стимулирует синтез липидов, обеспечивая получение для этого процесса необходимых субстратов (ацетил-КоА, глицерофосфат и NADPH2 ) из глюкозы. В жировой ткани инсулин тормозит мобилизацию липидов, что снижает концентрацию жирных кислот, циркулирующих в крови.
Влияние инсулина на метаболизм белков
Инсулин оказывает в целом анаболическое действие на белковый обмен. Он стимулирует потребление нейтральных аминокислот в мышцах и синтез белков в печени, мышцах и сердце.
Кроме того, инсулин регулирует клеточную дифференцировку, пролиферацию и трансформацию большого количества клеток. Инсулин поддерживает рост и репликацию многих клеток эпителиального происхождения, в том числе гепатоцитов, опухолевых клеток. Инсулин усиливает способность фактора роста фибробластов (ФРФ), тромбоцитарного фактора роста (ТФР), фактора роста эпидермиса (ФРЭ), простагландина (ПГF2 a), вазопрессина и аналогов цАМФ активировать размножение клеток.
Основные направления действия инсулина
1. Инсулин регулирует транспорт веществ
Инсулин стимулирует транспорт в клетку глюкозы, аминокислот, нуклеозидов, органического фосфата, ионов К+ и Са2+ . Эффект проявляются очень быстро, в течение нескольких секунд и минут.
Транспорт глюкозы в клетки происходит при участии ГЛЮТ. В мышцах и жировой ткани инсулинзависимый ГЛЮТ-4, в отсутствие инсулина находится в цитозольных везикулах. Под влиянием инсулина происходит транслокация везикул с ГЛЮТ в плазматическую мембрану и начинается транспорт глюкозы. При снижении концентрации инсулина, ГЛЮТ-4 возвращаются в цитозоль, и транспорт глюкозы прекращается.
2. Инсулин регулирует синтез ферментов
Инсулин влияет на скорость транскрипции более чем 100 специфических мРНК в печени, жировой ткани, скелетных мышцах и сердце. Эффект реализуется в течение несколько часов. В клетках печени инсулин индуцирует синтез ключевых ферментов гликолиза (глюкокиназы, фруктокиназы и пируваткиназы), ПФШ (глюкозо-6ф ДГ), липогенеза (цитратлиаза, пальмитатсинтаза, Ацетил-КоА-карбоксилаза), транспортеров глюкозы (?) и репрессирует синтез ключевого фермента глюконеогенеза (ФЕП карбоксикиназу).
3. Инсулин регулирует активность ферментов
Инсулин регулирует активность ферментов путем их фосфорилирования и дефосфорилирования. Эффект проявляются очень быстро, в течение нескольких секунд и минут.
· Инсулин активирует ключевые ферменты гликолиза: в печени, мышцах, жировой ткани – фосфофруктокиназу и пирруваткиназу; в печени – глюкокиназу; в мышцах - гексокиназу II.
· Инсулин ингибирует в печени глюкозо-6-фосфотазу, что тормозит глюконеогенез и выход глюкозы в кровь.
· Инсулин активирует фосфопротеинфосфотазу гликогенсинтазы и гликогенфосфорилазы, в результате активируется синтез гликогена и тормозится его распад.
· В адипоцитах инсулин активирует ключевой фермент липогенеза (АцетилКоА-карбоксилазу). Инсулин в гепатоцитах и адипоцитах активирует фосфопротеинфосфатазу, которая дефосфорилирует и инактивирует ТАГ-липазу, что тормозит липолиз.
· Инсулин снижает активность аминотрансфераз и ферментов цикла мочевины. Последний эффект инсулина характеризуется повышением активности РНК-полимеразы и концентрации РНК в печени. При этом увеличивается скорость образования полисом и рибосом.
· Инсулин активирует ФДЭ, которая снижает концентрацию цАМФ, прерывает эффекты контринсулярных гормонов: в печени и жировой ткани тормозит липолиз, в печени и мышцах - глюконеогенез.
МЕХАНИЗМ ДЕЙСТВИЯ ИНСУЛИНА
Инсулин связывается с инсулиновым рецептором (IR), находящимся на мембране. IR обнаружены почти во всех типах клеток, но больше всего их в гепатоцитах и клетках жировой ткани (концентрация достигает до 20000 на клетку). IR постоянно синтезируется (ген в 19 хромосоме) и разрушается. После связывания инсулина с IR весь комплекс погружается в цитоплазму, достигает лизосом, где инсулин разрушается, а IR может разрушаться, а может возвращаться мембрану. Т1/2 IR 7—12 ч, но в присутствии инсулина уменьшается до 2-3 ч.
При высокой концентрации инсулина в плазме крови, число IR может уменьшаться в результате усиленного разрушения в лизосомах. Также у IR может снижаться активность при его фосфорилировании по остаткам серина и треонина.
Рецептор инсулина ( IR) - гликопротеин, состоит из 2 α и 2 β субъединиц связанных дисульфидными связями. α субъединицы (719 АК) расположены вне клетки, они связывают инсулин, а β субъединицы (трансмебранный белок, 620 АК) обладают тирозинкиназной активностью. После присоединения гормона к α субъединицам, β субъединицы сначала фосфорилируют друг друга, а затем внутриклеточные белки — субстраты инсулинового рецептора (IRS). Известно несколько таких субстратов: IRS-1, IRS-2 (фосфопротеины, состоящие из более чем 1200 аминокислот), Shc, а также некоторые белки семейства STAT.
Активация инсулином сигнального пути Ras
Фосфорилированный инсулиновым рецептором She соединяется с небольшим цитозольным белком Grb. К образовавшемуся комплексу присоединяется с Ras-белок (из семейства малых ГТФ-связывающих белков, в неактивном состоянии прикреплён к внутренней поверхности плазматической мембраны и связан с ГДФ), GAP (от англ. GTP- ase activating factor — фактор, активирующий ГТФазу), GEF (от англ. GTP exchange factor — фактор обмена ГТФ) и SOS (от англ. son ofsevenless, названный по мутации гена у дрозофилы). Два последних белка способствуют отделению ГДФ от Ras-белка и присоединению к нему ГТФ, с образованием активной ГТФ-связанной формы Ras.
Активированный Ras соединяется с протеинкиназой Raf-1 и активирует ее в результате многоэтапного процесса. Активированная ПК Raf-1 стимулирует каскад реакций фосфорилирования и активации других протеинкиназ. ПК Raf-1 фосфорилирует и активирует киназу МАПК, которая, в свою очередь, фосфорилирует и активирует митогенактивируемые протеинкиназы МАПК.
МАПК фосфорилирует многие цитоплазматические белки: ПК pp90S6, белки рибосом, фосфолипазу А2 , активаторы транскрипции STAT.
В результате активации протеинкиназ происходит фосфорилирование ферментов и факторов транскрипции, что составляет основу многочисленных эффектов инсулина. Например:
Активация гликогенсинтазы
ПК pp90S6 фосфорилирует и активирует фосфопротеинфосфатазу (ФПФ). ФПФ дефосфорилирует и инактивирует киназу гликогенфосфорилазы и гликогенфосфорилазу, дефосфорилирует и активирует гликогенсинтазу. В результате активируется синтез гликогена, а распад - ингибируется.
Активация инозитолтрифосфатной системы
Фосфорилированные инсулином белки IRS-1 присоединяются к ФЛ С и активируют ее.
ФЛ С расщепляет фосфатидилинозитолы с образованием инозитолфосфатов и ДАГ.
Фосфорилированные инсулином белки IRS-1 и Shc присоединяются к фосфоинозитол-3-киназе (ФИ-3-киназа) и активируют ее.
ФИ-3-киназа катализирует фосфорилирование инозитолфосфатов (ФИ, ФИ-4-ф и ФИ-4,5-бф) в 3 положении, образуя инозитолполифосфаты: ФИ-3-ф, ФИ-3,4-бф, ФИ-3,4,5-тф. ФИ-3,4,5-тф (ИФ3 ) стимулирует мобилизацию Са2+ из ЭПР.
Са2+ и ДАГ активирует специфические ПК С.
Са2+ активирует микроканальцы, которые осуществляют транслокацию ГЛЮТ-4 в плазматическую мембрану, и таким образом ускоряет трансмембранный перенос глюкозы в клетки жировой и мышечной ткани.
Активация фосфодиэстеразы
Фосфорилированные инсулином белки IRS-1 и Shc присоединяются к протеинкиназе В (ПК В) и активируют ее. ПК В фосфорилирует и активирует фосфодиэстеразу (ФДЭ). ФДЭ катализирует превращение цАМФ в АМФ, прерывая эффекты контринсулярных гормонов, что приводит к торможению липолиза в жировой ткани, гликогенолиза в печени.
Регуляция транскрипции мРНК
STAT – особые белки, являются переносчиками сигнала и активаторами транскрипции. При фосфорилировании STAT с участием IR или МАПК образуют димеры, которые транспортируются в ядро, где связываются со специфическими участками ДНК, регулируют транскрипцию мРНК и биосинтез белков-фементов.
Путь Ras активируется не только инсулином, но и другими гормонами и факторами роста.
ГОСУДАРСТВЕННАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ
кафедра биохимии
Утверждаю
Зав. каф. проф., д.м.н.
Мещанинов В.Н.
_____‘’_____________2006 г
ЛЕКЦИЯ № 11
Тема: Сахарный диабет I и II типа: механизмы возникновения,
метаболические нарушения, осложнения.
Факультеты: лечебно-профилактический, медико-профилактический, педиатрический.
2 курс.
В норме уровень глюкозы в крови натощак составляет 3.3 – 5.5 ммоль/л.
Гипергликемия – повышение уровня глюкозы в крови выше 6,1 ммоль/л. Гипергликемия бывает физиологической и патологической.
Причины физиологической гипергликемии:
1) алиментарная, при употреблении легкоусвояемых углеводов. Не превышает 11 ммоль/л, нормализуется в течение 3 часов;
2) стрессорная , под действием катехоламинов, глюкокортикоидов, вазопрессина;
3) кратковременные физические нагрузки .
Причины патологической гипергликемии:
1) судороги при эпилепсиях, столбняке;
2) эндокринные нарушения . Гиперпродукция контринсулярных гормонов (гипертириоз, синдромы Кушинга и Кона), абсолютный или относительный дефицит инсулина (сахарный диабет).
3) ЧМТ .
Гипогликемия снижение уровня глюкозы в крови ниже 3,3 ммоль/л. Гипогликемия бывает физиологической и патологической.
Причины физиологической гипогликемии: 1) алиментарная, при голодании; 2) длительная физическая нагрузка .
Причины патологической гипогликемии: 1) эндокринные нарушения при избытке инсулина (инсулинома – доброкачественная опухоль β-клеток, передозировка инсулина у больных СД) или недостаточности контринсулярных гормонов (гипотиреоз, дефицит глюкокортикоидов); 2) гликогенозы, агликогенозы, препятствующие гликогенолизу; 3) печеночная недостаточность, связанная с низкой активностью глюконеогенеза; 4) почечная недостаточность, связанная с врожденной патологией реабсорбции глюкозы (почечный диабет); 5) отравления монойодацетатом (вызывает глюкозурию).
Сахарный диабет (СД) — системное гетерогенное заболевание, обусловленное абсолютным или относительным дефицитом инулина, который сначала вызывает нарушение углеводного, а затем всех видов обмена, что в итоге поражает все функциональные системы организма.
СД широко распространенное заболевание, им страдает 6,6% населения, в России – 5%.
СД бывает первичным и вторичным. Кроме того, выделяют нарушение толерантности к глюкозе и СД беременных.
Первичный СД - самостоятельное заболевание.
Вторичный СД является симптоматическим, он возникает при патологии эндокринных желез (акромегалия, феохромоцитома, глюкагонома, синдромы Кушинга, Кона) и патологии поджелудочной железы (хронический панкреатит, рак, панкреатэктомия, гемохроматоз, генетические синдромы).
Первичный СД по механизму развития подразделяется на СД I типа (раньше ИЗСД) и СД II типа (раньше ИНСД).
Общими симптомами любого СД являются жажда, полиурия, кожный зуд, склонность к инфекциям.
Этиологическая классификация СД (ВОЗ 1999) .
1. Сахарный диабет I типа (раньше ИЗСД)
а). Аутоиммунный
б). Идиопатический
2. СД II типа (раньше ИНСД)
3. Другие специфические типы
а). генетические дефекты β-клеток
б). генетические дефекты в действии инсулина
в). болезни экзокринной части поджелудочной железы (панкреатит и т.д.)
г). эндокринопатии
д). СД, индуцированный лекарствами и химикатами (глюкокортикоиды, никотиновая кислота, тиреоидные гормоны, тиазиды, вакор, пентамидин и т.д.)
е). Инфекции (врожденная краснуха, цитомегаловирус и т.д.).
ж). Необычные формы иммуноопосредованного диабета.
з). Другие генетические синдромы, иногда сочетающиеся с диабетом (Дауна, Тернера и т.д.).
4. Гестационный СД (беременных)
САХАРНЫЙ ДИАБЕТ I типа
СД I типа — заболевание, которое возникает вследствие абсолютного дефицита инсулина, вызванного аутоиммунным разрушением β-клеток поджелудочной железы. СД I типа поражает в большинстве случаев детей, подростков и молодых людей до 30 лет, но может проявиться в любом возрасте. СД I типа редко является семейным заболеванием (10-15% всех случаев).
Причины СД I типа
1. Генетическая предрасположенность . Генетические дефекты ведущие к СД могут реализоваться в клетках иммунной системы и β-клетках поджелудочной железы. В β-клетках известно около 20 генов, способствующих развитию СД I типа. В 60-70% случаях СД I типа связан с наличием в 6 хромосоме HLA региона генов DR3, DR4 и DQ.
2. Действие на β-клетки β-цитотропных вирусов (оспа, краснуха, корь, паротит, Коксаки, аденовирус, цитомегаловирус), химических и других диабетогенов .
Вариант 1
При наличии генетического дефекта, на поверхности β-клеток накапливаются антигены, имеющие схожую аминокислотную последовательность с β-цитотропными вирусами.
В случае возникновения инфекции β-цитотропных вирусов, развиваются иммунные реакции против этих вирусов и аутоиммунные реакции против схожих антигенов β-клеток. Реакция идет с участием моноцитов, Т-лимфоцитов, антител к β-клеткам, инсулину, глутамат декарбоксилазе (фермент 64кДа, находиться на мембране β-клеток). В результате аутоиммунные реакции вызывают гибель β-клеток.
Вариант 2
При действии на β-клетки с генотипом HLA β-цитотропных вирусов или диабетогенов на поверхности β-клеток происходит изменение антигенов.
На измененные антигены β-клетки развиваются аутоиммунные реакции. Аутоиммунные реакции вызывают гибель β-клеток.
Вариант 3
β-цитотропные вирусы имеют схожую последовательность аминокислот с глутамат декарбоксилазой β-клеток. Генетический дефект СД8+ лимфоцитов (Т-супрессоров) не позволяет им отличить аминокислотную последовательность вируса и глутамат декарбоксилазы, поэтому при возникновении инфекции, Т-лимфоциты реагируют на глутамат декарбоксилазу β-клеток как на вирус.
Вариант 4
Некоторые β-цитотропные вирусы и химические диабетогены, например, производные нитрозомочевины, нитрозамины, аллоксан самостоятельно и избирательно поражают β-клетки, вызывая их лизис;
Стадии развития СД I типа
1. Стадия генетической предрасположенности . Есть генетические маркеры, нет нарушений углеводного обмена. Может длиться всю жизнь;
2. Стадия провоцирующих событий . Инфекция β-цитотропных вирусов или действие химических диабетогенов. Протекает без клинических симптомов;
3. Стадия явных иммунных аномалий . Развитие смешанных аутоиммунных реакций против β-клеток. Ресурсы инсулина достаточны. Протекает без клинических симптомов. Развивается от 2-3 месяцев до 2-3 лет;
4. Стадия латентного диабета . Гибель 75% β-клеток, небольшое снижение инсулина, гипергликемия при нагрузочных пробах, снижение аутоиммунных процессов. Протекает без клинических симптомов;
5. Явный диабет . Гибель 80-90% β-клеток, заметное снижение инсулина, гипергликемия натощак, нет или слабые аутоиммунные реакции. Появляются клинические симптомы. Развивается 2 года. Необходима инсулинотерапия;
6. Терминальный диабет . Полная гибель β-клеток, высокая потребность в инсулинотерапии, аутоиммунные проявления снижены или их нет. Выраженные клинические проявления, появляются ангиопатии. Развивается до 3,5 лет;
Изменения метаболизма при СД I типа
При СД I типа исчезает инсулин, т.к. инсулин ингибитор секреции глюкагона, в крови происходит увеличение глюкагона.
Изменения в углеводном обмене
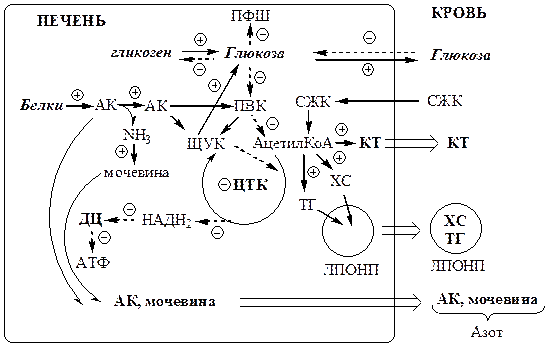
В печени дефицит инсулина и избыток глюкагона стимулирует реакции глюконеогенеза, гликогенолиза и ингибирует реакции гликолиза, ПФШ и синтеза гликогена. В результате в печени глюкозы больше образуется, чем потребляется.
Так как реакции глюконеогенеза протекают через ЩУК, он, образовавшись из ПВК, аспартата и малата, активно вовлекается в глюконеогенез, вместо того чтобы включаться в ЦТК. В результате ЦТК и ДЦ тормозится, снижается образование АТФ, возникает энергодефицит .
В инсулинзависимых тканях (мышцы, жировая ткань) дефицит инсулина препятствует поступлению глюкозы в клетки и ее использованию в реакциях гликолиза, ПФШ и синтеза гликогена. Блокирование ЦТК и ДЦ также вызывает энергодефицит.
Снижение потребления глюкозы инсулинзависимыми тканями и усиление ее образования в печени приводит к гипергликемии . Когда гипергликемия превышает концентрационный почечный порог возникает глюкозурия.
Глюкозурия – наличие глюкозы моче. В норме проксимальные канальцы почек реабсорбируют всю фильтрующуюся в клубочках глюкозу. Если уровень глюкозы превышает в крови 9-10 ммоль/л, глюкоза не успевает полностью реабсорбироваться из первичной мочи и частично выводится с вторичной мочой.
У больных с СД после приёма пищи концентрация глюкозы в крови может достигать 300-500 мг/дл и сохраняется на высоком уровне в постабсорбтивном периоде, т.е. снижается толерантность к глюкозе.
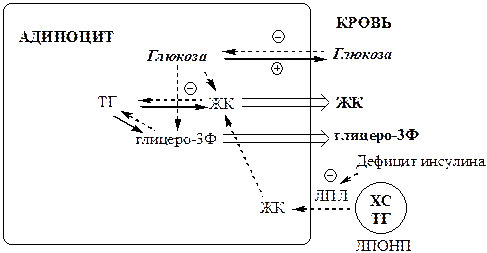
Изменения в липидном обмене
9-09-2015, 00:12