У підшлунковій залозі синтезується секреторний панкреатичний інгібітор трипсину. Дуже важливим захисним механізмом є здатність панкреатичних ферментів до аутолізу, самоперетравлення. Справа в тім, що багато ферментів підшлункової залози, перш за все трипсин, здатні розщеплювати самі себе, обмежуючи процес власної активації. Крім того, в печінці утворюються білки: α1 -антитрипсин і β2 -макроглобулін, які зв’язують активні панкреатичні ферменти, якщо вони потрапляють в кров або в перитонеальну рідину.
Трипсини є досить складною молекулою, що складається з двох субодиниць. Сполучаючись одна з одною, ці субодиниці утворюють центр розпізнавання амінокислот, який знаходиться у вузькій щілині між субодиницями. Центр розпізнає дві амінокислоти: аргінін та лізин в молекулі субстрату, і в цьому місці трипсин розщеплює поліпептидний ланцюг.
Дві субодиниці трипсину зв’язуються між собою за допомогою одного амінокислотного ланцюга, який знаходиться на протилежній від центру розпізнавання стороні молекули. Панкреатичний інгібітор трипсину проникає між двома субодиницями трипсину і блокує активний центр молекули. Встановлено, що на амінокислотному ланцюгу, який з’єднує субодиниці в положенні 117, знаходиться аргінін. При аутолізі саме ця амінокислота піддається розчепленню самим трипсином та іншими трипсиноподібними ферментами. В результаті відбувається роз’єднання субодиниць і руйнування активного центру.
Таким чином, можна визначити два основних механізми, що стримують активацію трипсину (мал. 4). Перший – інгібітор трипсину, що блокує активний центр, але він здатний нейтралізувати лише 20 % від всієї кількості трипсину, який може утворюватись в ацинарній клітині. Другий механізм – сам трипсин та інші протеолітичні ферменти, що здатні розщеплювати молекулу трипсину в положенні 117, що призводить до втрати активності цього ферменту.
При спадковому панкреатиті аргінін в положенні 117 замінюється на гістидин, тобто на амінокислоту, яку не здатен розпізнавати і, відповідно, розщепляти активний центр молекули трипсину. Це призводить до того, що трипсин та подібні до нього ферменти втрачають контроль над власною активацією. Вони вже не здатні пригнічувати трипсиноген, активний трипсин, і єдиною ланкою захисту залишається панкреатичний інгібітор трипсину.
Таким чином, якщо при спадковому панкреатиті одночасно відбувається активація великої кількості трипсину, кількості більшої, ніж здатен заблокувати панкреатичний інгібітор, це призводить до неконтрольованої активації трипсину. В свою чергу він активує інші ферменти панкреатичного каскаду, і це буде виявлятися у вигляді нападу гострого панкреатиту.
В останні роки вивчалася частота генетичних мутацій при захворюваннях підшлункової залози. Так, була досліджена велика група пацієнтів, яким проводилась ендоскопічна ретроградна холангіопанкреатографія. Із 102 хворих у 55 (54 %) встановлений діагноз так званого ідіопатичного панкреатиту, тобто етіологія хвороби не була з’ясована. У цих хворих досліджувались основні відомі генетичні мутації, як могли привести до пошкодження підшлункової залози: мутація регулятора трансмембранної проникливості, що зумовлювала виникнення муковісцидозу, мутація, що призводила до виникнення дефіциту α1 -антитрипсину, і мутація спадкового панкреатиту із зміною молекули трипсиногену в положенні 117.
Встановлено, що у 15 (14,7 %) із 102 хворих були наявні генетичні мутації: у 2 виявлено мутацію спадкового панкреатиту, у 6 – гетерозиготна мутація гену муковісцидозу, у 1 – гомозиготна мутація, що призводила до дефіциту α1 -антитрипсину, та у 6 – гетерозиготність по цій мутації.
Подальше вивчення сім’ї S., в якій D.C.Whitcomb виявив мутацію R117, показало, що 12,5 % хворих спадковим панкреатитом, в яких є усі класичні діагностичні його ознаки, чіткі ознаки самого панкреатиту та спадковий характер цієї хвороби, не мають мутації R117.
Дослідження показали, що існують і інші мутації, які призводять до виникнення цього захворювання. Так, французькі автори, які вивчали 14 родин, хворих на спадковий панкреатит, виявили мутацію R117 в 4 родинах, в той час як у 3 сім’ях була виявлена мутація в іншому, 2-му екзоні: мутація К18R (заміна лізину на аргінін в положенні 18) і N241. Сам D.C.Whitcomb виявив ще одну мутацію в положенні N211, за якої спостерігався більш пізній початок захворювання та м’яке протікання панкреатиту.
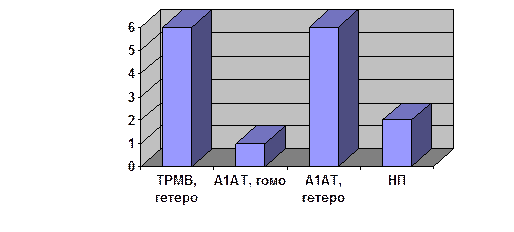
Мал 2. Частотність генетичних мутацій у хворих панкреатитом: генетичні мутації виявлені в 14,7 % хворих (за C . P . Choudari et al ., 1998)
У чому ж полягає клінічне значення нових даних про патогенез спадкового панкреатиту? Якщо говорити про власне спадковий панкреатит, то отримані результати пояснюють аутосомно-домінантний характер успадкування цього захворювання. Тобто для виникнення спадкового панкреатиту досить, щоб хоча б половини молекули трипсину була стійкою до гідролізу. З іншого боку, ця теорії пояснює різну експресивність спадкового панкреатиту та інтермітуюче протікання цієї патології, тобто напад гострого панкреатиту виникатиме тоді, коли проходить внутрішньоклітинна активація великої кількості молекул трипсину.
Подальші дослідження сім’ї S. виявили цікаву закономірність. Згідно класичним діагностичним ознакам спадкового панкреатиту, діагноз встановлювався значною мірою на основі виключення інших причин панкреатиту — перш за все зловживання алкоголем і жовчнокам’яної хвороби. В цей же час при дослідженні цієї сім’ї (однієї з найбільш крупних) було виявлено, що серед хворих спадковим панкреатитом достовірно вищий відсоток осіб, які зловживали алкоголем, і достовірно вища частота проведення холецистектомій, ніж серед здорових членів цієї родини (мал. 3).

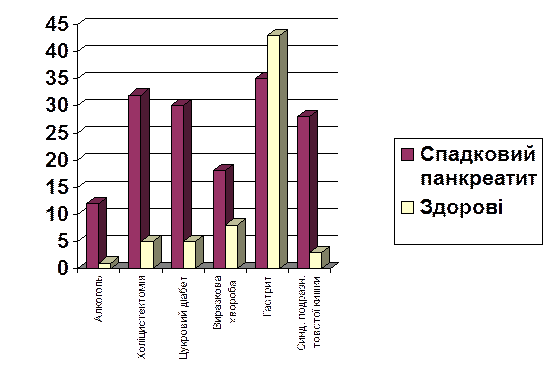
Мал 3. Порівняльна характеристика хворих спадковим панкреатитом і здорових в сім’ї S .
Це говорить про те, що зміна трипсину є лише тлом. Мутація призводить не до посилення активації трипсину, а лише порушує один із захисних механізмів ацинарних клітин. Для виникнення захворювання, появи клінічних симптомів необхідні преципітуючі фактори, які викликали б цю посилену активацію.
Таким чином, не можна вважати зловживання алкоголем і наявність холелітіаза факторами, що виключають спадковий панкреатит. Для верифікації діагноза необхідне виявлення катіонного трипсиногену за допомогою полімеразної ланцюгової реакції.
Генна теорія якоюсь мірою пояснює патогенез гострого панкреатиту в цілому. Зокрема, вона відкриває новий захисний механізм: аутоліз молекули трипсину . Генна теорія показує, що раннім пусковим механізмом гострого панкреатиту може бути внутрішньоклітинна активація трипсину. Це повертає нас до теорії самоперетравлювання підшлункової залози, яку висунув Chiariбільше 100 років тому, що показує основну роль трипсину в пошкодженні підшлункової залози. Згідно теорії, гострий панкреатит принаймні в частині випадків може починатися всередині ацинарної клітини.
Спадковий панкреатит при тривалому лікуванні (більше 10-15 років) призводить до виникнення хронічного панкреатиту . Морфологічні дослідження тканини підшлункової залози показали, що гістологічна картина при цьому захворювання нічим не відрізняється від гістологічної картини хронічного алкогольного панкреатиту. Це дозволяє припустити, що рецидивуючий гострий панкреатит може призводити до виникнення хронічного панкреатиту.
Запалення підшлункової залози може протікати субклінічно, якщо надлишкова активація трипсину проходить одночасно лише в невеликій кількості ацинарних клітин. Це може призвести до того, що не виявляються фізикальні, лабораторні чи інструментальні ознаки запалення підшлункової залози. Зміни протоків, які спостерігаються при хронічному спадковому панкреатиті, можуть бути проявами цього захворювання, а не причиною його.
Відомо, що спадковий панкреатит різко підвищує ризик розвитку раку підшлункової залози. При дослідженні хворих спадковим панкреатитом з 21 родини рак підшлункової залози був виявлений у 15 %. У 9 % хворих виявлені пухлини черевної порожнини позапанкреатичної локалізації. Встановлено, що у хворих спадковим панкреатитом до 70 років ризик виникнення раку складає 40 %.
У той же час при генетичних дослідженнях хворих спадковим панкреатитом змін онкогенів чи генів-супресорів пухлинного росту не виявлено. Це дозволяє припустити, що причина вищої частоти раку при спадковому панкреатиті полягає у вищому рівні активності запального процесу в підшлунковій залозі при спадковому панкреатиті й більшою тривалістю запального процесу. Це зайвий доказ на користь того, що хронічне запалення є фактором, що сприяє розвитку раку підшлункової залози.
СПИСОК ВИКОРИСТАНОЇ ЛІТЕРАТУРИ
1. Острый панкреатит // Международный медицинский журнал. – Т.5. - № 2. – 1999. – С. 87-91.
2. Иннервация поджелудочной железы // Российский журнал гастроэнтерологии, гепатологии, колопроктологии. – Т.6. - № 1. – 1996. – С. 35-39.
3. Коротько Г.Ф. Регуляция секреции поджелудочной железы // Российский журнал гастроэнтерологии, гепатологии, колопроктологии. – Т.9. - № 4. – 1996. – С. 6-9.
4. Буеверов А.О. Медиаторы воспаления и поражение поджелудочной железы // Российский журнал гастроэнтерологии, гепатологии, колопроктологии. – Т.9. - № 4. – 1996. – С. 15-18.
5. Охлобыстин А.В. Новые данные о патогенезе наследственного панкреатита // Российский журнал гастроэнтерологии, гепатологии, колопроктологии. – Т.9. - № 4. – 1996. – С. 18-23.
6. Учебно-методическая разработка для студентов VI курса по теме: «Острый панкреатит» / Сост. Почечуев Е.А. и др. – М.: Изд-во МГМУ, 1995.
7. Розин Д.Г. Ферментовыделительная деятельность поджелудочной железы. – Ташкент: Медицина, 1981. – С. 164.
8-09-2015, 21:08