Перші ознаки включення компенсаторно-пристосувальних процесів в міокарді спостерігаються вже через 35 діб після тиреоїдектомії, проте на цьому етапі вони існують разом із реактивними, а часто і деструктивними процесами, що свідчить про їх невисоку ефективність. Найбільш вираженими процеси адаптації стають через 50 діб після операції. Причому, виразність цих процесів була більшою в міокарді правого передсердя, в той час як в міокарді лівого шлуночка поряд з компенсаторно-пристосувальними, все ще спостерігались і деструктивні ознаки. В цей час відбувається посилення активності НАДФН+ -залежного шляху перекисного окислення ліпідів (концентрація малонового діальдегіду становила (125,5 ± 3,4) ммоль/г при (54+ 6,9) – у контролі). Це опосередковано свідчить про збільшення вмісту в кардіоміоцитах відновленої форми НАДФ, а отже і про посилення її утворення в дихальному ланцюгу мітохондрій [Афонина Г. Б., 2000]. Разом з тим, в структурі ліпідів міокарда підвищується вміст поліненасичених жирних кислот, що збільшує стійкість мембран кардіоміоцитів та сприятливо впливає на процеси трансмембранного транспорту і рецепції. З іншого боку, на 9,23% зменшився вміст насичених жирних кислот в складі ліпідів міокарда. Причиною таких змін може бути посилене їх використання для процесів окислення в мітохондріях кардіоміоцитів, де вони є основним джерелом АТФ [Хехт А., 1975; Галявич А. С., 2006]. Варто зазначити, що показник осмотичної резистентності еритроцитів в цій стадії залишається низьким.
В ендотелії капілярів виявлені ознаки помірної активності як транспортних, так і біосинтетичних процесів. Це сприяє відновленню мікроциркуляції в них, оптимізації обмінних процесів, що разом із зменшенням ітерстиційного набряку покращує трофіку кардіоміоцитів, без чого неможливим є становлення компенсаторних механізмів в них.
В кардіоміоцитах найбільш вираженими виявились компенсаторні реакції білоксинтетичного апарату. Вони були типовими для міокарда [Меерсон Ф. З., 1980; Непомнящих Л. М., 1991; Пауков В. С., 1982] та полягали в гіпертрофії ядер, збільшенні кількості вільних рибосом, полісом та зв’язаних з мембранами гранулярної ендоплазматичної сітки форм. Показник об’ємної щільності міофібрил не змінився, проте їх якісні характеристики відображали активні репаративні процеси. Ознакою останніх є наявність довгих ланцюжків полісом, на яких синтезуються актинові та міозинові міофіламенти. Проте, як відомо, за умови гіпотиреозу переважно синтезуються в-ізоформа важкого ланцюга міозину, потужність та швидкість скорочення для якої нижча, порівняно з б-ізоформою [Morkin Е., 1993; OjamaaK., 1996; LadensonP. W., 1992]. Компенсаторно-пристосувальні процеси мітохондрій полягали переважно у вираженій їх гіперплазії, внаслідок якої утворювались молоді форми органел (рис. 2). А в міокарді правого передсердя, спостерігалась і гіпертрофія окремих мітохондрій. Разом з тим, їх ультраструктури відновлюються лише частково. На цьому етапі відбувається компенсація енергетичного дефіциту в кардіоміоцитах як за рахунок мітохондріальних механізмів, так і за рахунок активізації процесів гліколізу. Це узгоджується з даними гістохімічних досліджень в динаміці гіпотиреозу [Павлюк В. Г., 1977; Колесова Н. А., 2007]. Клітини, які не змогли переключитись на гліколітичний шлях отримання енергії, зазнають загибелі. Про це свідчить наявність значної кількості гранул глікогену в поодиноких змінених за „темним” типом кардіоміоцитах. В кардіоміоцитах все більше виникає ознак порушення інтрацелюлярного обміну кальцію, що відображено в особливостях будови саркоплазматичної сітки, накопиченні електронно-щільних депозитів в матриксі мітохондрій, контрактурних змінах міофібрил.
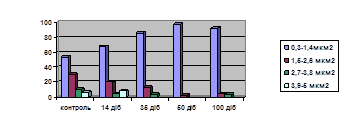
Рис. 2. Розподіл мітохондрій в кардіоміоцитах лівого шлуночка міокарду інтактних щурів та в динаміці експериментального гіпотиреозу. По осі абсцис - групи досліджень, по осі ординат – кількість досліджених об’єктів в %.
Ймовірно, ця стадія розвитку міокардіопатії є ще оборотною та сприятлива для будь-якої медикаментозної корекції (етіопатогенетичного лікування гіпотиреозу або симптоматичної терапії проявів міокардіопатії). Після неї формується стадія декомпенсації міокарда з ознаками розвитку хронічної серцевої недостатності, які є необоротними. В нашому дослідженні ці зміни спостерігались через 100 діб після тиреоїдектомії. Вони супроводжувались зниженням активності НАДФН+ -індукованого перекисного окислення ліпідів. Це опосередковано вказує на вторинне порушення окисно-відновних процесів в мітохондріях. Активність спонтанного утворення продуктів перекисного окислення ліпідів все ще залишалась стабільно високою і становила (56,0 ± 10,0) ммоль/г при контрольному значенні - (26+ 4,2) ммоль/г. З ними, очевидно, пов’язані зміни в жирнокислотному спектрі ліпідів міокарду. На фоні відновлення співвідношення насичених і ненасичених жирних кислот, в структурі останніх зменшується вміст поліненасичених жирних кислот, які, як відомо, першими включаються в процес перекисного окислення. Як наслідок - знову, вторинно, порушуються проникність, трансдукція та ригідність мембран кардіоміоцитів. Подібне співвідношення жирних кислот в структурі ліпідів крові спостерігали і при інших хронічних захворюваннях міокарду у людини [Алимова Е. К., 1970; Pirro М., 2002]. Структурна перебудова клітинних мембран сприяє зменшенню її міцності та проникності, що корелює із зниженням осмотичної резистентності еритроцитів більше, ніж на 200%.
Зрив адаптаційних процесів проявився посиленням деструктивних змін в першу чергу з боку скоротливого та енергетичного апаратів на фоні вираженої недостатності репаративних процесів, що проявляється утворенням глибоких інвагінацій каріолеми з формуванням сегментованих ядер; розширенням перинуклеарного простору, зменшенням вмісту вільних рибосом, полісом та канальців гранулярної ендоплазматичної сітки. Наслідком гіпопластичних та активації деструктивних процесів в міокарді є зменшення вмісту міофібрил, структурна цілісність яких в різній мірі порушена, гетерогенність змін в мітохондріях.
Ми спостерігали початкові етапи апоптозу у вигляді перескорочених і «темних» клітин, проте нам не вдалось виявити кардіоміоцити на термінальних стадіях процесу. Можливо, цей процес не завершувався і високий внутрішній антиапоптичний потенціал дозволяв тривалий час підтримувати життєдіяльність кардіоміоцитів у вигляді їх гібернації, що часто спостерігається при різних захворюваннях міокарда [Brown Т. А., 2001; SchulzR., 2000 ]. За умови енергетичного дефіциту в клітині, на початкових стадіях процес апоптозу міг завершуватись розвитком некротичних змін в кардіоміоцитах, які мали місце в цей період дослідження.
Очевидними були механізми порушення внутрішньоклітинного обміну кальцію, які прогресували в динаміці експерименту. Вони проявились колапсом канальців саркоплазматичної сітки, відкладанням електронно щільних депозитів в мітохондріях та ділянками перескорочення міофібрил.
З розвитком декомпенсації поновлюється активація колагеноутворення фібробластами, що спостерігалось на етапі реактивних змін; відбувається синтез значної кількості еластичних волокон, які відкладаються як периваскулярно, так і в інтерстиційному просторі; організація сполучною тканиною ділянок мікрогеморагій в міокард. Таким чином, через 100 діб після тиреоїдектомії формуються ознаки дифузного дрібновогнищевого склерозу та фіброзу [Гавриш А. С., 2007].
Стадія декомпенсації міокарда при гіпотиреозі характеризується класичними електрокардіографічними ознаками – синусовою брадикардією, подовженням інтервалу РQ та зниженням амплітуди всіх зубців на електрокардіограмі [Левина Л. И., 1989]. Ці дані відображають зниження біоелектричної активності провідної системи серця та розвиток дисметаболічних процесів в ньому. Виявлені нами зміни свідчать про етапність розвитку гіпотиреоїдної міокардіопатії, результатом якої є розвиток хронічної серцево-судинної недостатності.
Морфофункціональний аналіз стану міокарда щурів при корекції гіпотиреозу L-тироксином. Ґрунтуючись на отриманих даних щодо концентрації вільного тироксину в плазмі крові, можна стверджувати про адекватність підібраної нами дози L-тироксину (10 мкг/кг), оскільки у щурів мав місце стан еутиреозу, хоча і спостерігалась індивідуальна різниця концентрації вільного тироксину, яку можна пов’язати з особливостями фармакокінетики препарату (рис. 1). Звертає на себе увагу не лише статистично достовірне зниження рівня загального кальцію в плазмі крові тварин, яке також характерне для нелікованого гіпотиреозу, а і іонізованої його форми.
Проведені нами в комплексі морфологічні, біохімічні та фізіологічні дослідження дають право стверджувати про те, що лікування післяопераційного гіпотиреозу L-тироксином не запобігає розвитку гіпотиреоїдної міокардіопатії як в міокарді правого передсердя, так і лівого шлуночка. Проте, воно здатне відстрочити її формування в часі. Так, виявлені нами біохімічні, фізіологічні та морфологічні ознаки реактивних процесів в міокарді нелікованих тварин через 14 діб після тиреоїдектомії, найбільш виразно проявлялись лише через 35 діб в групі лікованих L-тироксином тиреоїдектомованих щурів. Компенсаторно-пристосувальні процеси зазнавали свого становлення через 50 діб і були добре виражені як в міокарді правого передсердя, так і лівого шлуночка. Перші ознаки декомпенсації з’являлись через 100 діб, проте вони були менш виражені, ніж у відповідний термін без лікування, тобто цей процес затримується в часі.
Разом із загальними ознаками гіпотиреоїдної міокардіопатії, на яких ми ґрунтувались для встановлення стадії процесу, в міокарді лікованих тироксином щурів виникають специфічні зміни, пов’язані з впливом екзогенного L-тироксину. В першу чергу це стосується міокарду правого передсердя. Цікаво, що в передсердних кардіоміоцитах тиреоїдектомованих щурів під впливом L-тироксину відбувалось утворення типових тріад, не характерних для цього відділу серця. Така перебудова передсердних кардіоміоцитів, очевидно, пов’язана з гіпертрофією міокарду передсердя та наростанням в передсердних кардіоміоцитах скоротливої їх функції. Ознаки гіпертрофії міокарда правого передсердя встановлені електрокардіографічно, що супроводжується формуванням вираженого р-«pulmonale». Утворення тріад можна розглядати як компенсаторну реакцію клітини на надлишок іонів кальцію в саркоплазмі. На користь участі кальцієвих механізмів пошкодження кардіоміоцитів свідчать електроннощільні депозити в матрикс мітохондрій, ущільнення кортикального шару саркоплазми та наявність численних перескорочених та «темних» клітин.
Виявлені нами розходження та набряк вставних дисків свідчать про порушення провідності між кардіоміоцитами. Вони з’являються в експерименті в стадії декомпенсації та, очевидно, посилюють прояви серцевої недостатності.
Іншою особливістю впливу L-тироксину на міокард тиреоїдектомованих щурів є активація всіх білоксинтетичних процесів як в кардіоміоцитах, так і в ендотеліоцитах та клітинах сполучної тканини. Якщо в кардіоміоцитах та ендотеліоцитах активація білкового синтезу посилює репараційні та обмінні процеси, відстрочуючи тим самим розвиток хронічної серцевої недостатності, то посилення колагеноутворення фібробластами, навпаки, сприяє формуванню вираженого міокардіофіброза, за рахунок якого відбувається порушення проникності гістогематичного бар’єру міокарда під час зриву адаптаційних процесів, що сприяє формуванню вторинних дистрофічних змін в ньому.
Характеристика міокарду при комбінованій корекції гіпотиреозу L-тироксином та кальцитоніном та після трансплантації фетальних щитоподібних залоз. Виявлена нами недостатність монотерапії гіпотиреоїдної міокардіопатії L-тироксином може бути обумовлена дефіцитом іншого гормону щитовидної залози – кальцитоніном. Відомо, що вміст цього гормону знижується у людей з післяопераційним гіпотиреозом [Schneider Р., 1991; Kouneva-SkerlevaV. H., 2006]. Використання кальцитоніну при комбінованому з тироксином лікуванні забезпечує більшу збереженість структур міокарда, ніж при застосуванні єдиного L-тироксину. Це супроводжується адекватним заміщенням тироксину в плазмі крові (рис. 1), а також відновленням рівнів загального та іонізованого кальцію крові до контрольних значень. Складний каскад вторинних посередників, що запускаються рецепторно-гормональною взаємодією чинить значну кількість ефектів, регулюючи проникність іонних каналів в кардіоміоцитах, процеси диференціювання та клітинної загибелі [FindlayD. M., 2002; SueurS., 2005]. Можливо саме з антиапоптичним ефектом цього гормону пов’язане те, що вміст «темних» та контрактурно-змінених кардіоміоцитів при його застосуванні на пізніх термінах післяопераційного гіпотиреозу був незначним, а через 100 діб взагалі не виявлялись перескорочені кардіоміоцити.
Оскільки відомо, що кальцитонін регулює проникність мембран кардіоміоцитів для кальцію, то очевидним є його вплив на регуляцію концентрації Са2+ в саркоплазмі [BickR. J., 2005; SainiH. K., 2007]. Схоже, точкою прикладання цього гормону є мембрани канальців саркоплазматичної сітки, де він відновлює активність Са2+ -АТФази та надходження іонів кальцію з саркоплазми в середину канальців. Підтвердженням такої думки може бути структурна цілісність саркоплазматичної сітки, відсутність ознак кальцієвих пошкоджень в кардіоміоцитах як правого передсердя, так і лівого шлуночка на пізніх термінах експерименту. Ми не виявили ознак міокардіофіброза при комбінованому з кальцитоніном лікуванні, які є проявом хронічної серцевої недостатності. Це можна пов’язати з позитивним впливом гормону на процеси ремоделювання в сполучній тканині. Використання трансплантації фетальних щитоподібних залоз дало тривалий ефект лікування. При цьому через 100 діб досягався рівень еутиреозу (рис. 1), нормалізувались показники обміну кальцію в організмі, що є ознакою функціональної активності трансплантатів. Вражала збереженість ультраструктури кардіоміоцитів як в міокарді правого передсердя, так і лівого шлуночка. Проте, нами встановлені деякі особливості переважно з боку сполучної тканини міокарду. Окрім помірно вираженого інтерстиційного набряку та формування дифузного міокардіофіброзу, в периваскулярному просторі виявлені низькодиференційовані, з високим ядерно-цитоплазматичним співвідношенням, клітини, в цитоплазмі яких містились поодинокі гранули, що містили значно гіпертрофовані ядерця із переважанням фібрилярного компоненту, гігантські мітохондрії. В ендотеліоцитах спостерігались мікроядра. Такі зміни являють собою ознаки клітинного атипізму, що, в свою чергу, з часом може призвести до пухлинного переродження таких клітин [Благодаров В. М., 1997; Струков А. И., 1995]. Отримані дані ставлять перед нами питання щодо доцільності використання трансплантації фетальних щитовидних залоз з метою корекції післяопераційного гіпотиреозу, враховуючи її корисний ефект та можливий шкідливий вплив для організму. Комплексна оцінка ендокринного апарату серця в динаміці гіпотиреозу та при його корекції. З огляду на доведену участь передсердного натрійуретичного пептиду (ПНУП) в розвитку серцевої недостатності [Поливода С. Н., 2004; Скворцов А. А., 2003; EllisR., 1998], варто детальніше зупинитись на ендокринній функції серця. Встановлено, що в динаміці гіпотиреозу першими уражаються механізми виділення гормону з кардіоміоцитів, а його синтез порушується лише в стадії декомпенсації. Це супроводжується ознаками гіпотрофії білоксинтетичного апарату в передсердних кардіоміоцитах та характерним перерозподілом типів гранул, що відображає зниження вмісту новоутворених форм та накопичення безмембранних гранул в саркоплазмі (рис. 3). Майже вдвічі зменшується показник середньої площі всіх типів гранул. Виявлені ознаки порушення базальної секреції ПНУПу. В якості компенсаторного механізму, активується стимульована секреція недозрілих гранул, про що дає право думати їх екзоцитоз з кардіоміоцитів та поява поодиноких гранул в інтерстиційному просторі на пізніх етапах гіпотиреоза. Підвищення концентрації іонів Са2+ в саркоплазмі сприяє екзоцитозу даного пулу мембрановмісних гранул, вкритих клатрином, використовуючи G-білки [BensimonM., 2005; KleinR. M., 1993]. Вміст частини гранул організується з фібрилярними білками та обмежовується мембранами комплексу Гольджі, переходячи в пул мембранних гранул. Частина безмембранних гранул зливається з лізосомами, ферменти яких, очевидно, беруть участь в елімінації ПНУП. Використання з терапевтичною метою L-тироксину стимулює процеси білкового синтезу, в тому числі і ПНУПу в передсердних кардіоміоцитах, що сприяє врівноваженню вмісту різних типів передсердних гранул, проте не попереджає накопиченню безмембранних форм гранул (рис. 3). Кальцитонін (екзогенний або той, що походить з трансплантованих фетальних щитоподібних залоз), відновлюючи внутрішньоклітинний обмін іонів кальцію сприяє врівноваженню процесів синтезу, депонування, дозрівання та секреції гормону на пізніх етапах експерименту, що, очевидно, відіграє важливу роль в попередженні розвитку хронічної серцевої недостатності та артеріальної гіпертензії (рис. 3).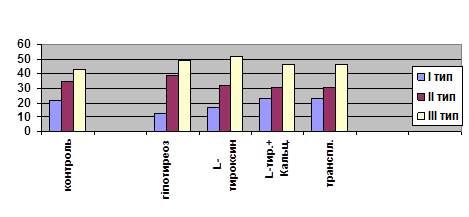 Рис. 3. Розподіл передсердних гранул в дослідних групах тварин через 100 діб після операції. По осі абсцис - групи досліджень, по осі ординат – кількість досліджених об’єктів в %.
Рис. 3. Розподіл передсердних гранул в дослідних групах тварин через 100 діб після операції. По осі абсцис - групи досліджень, по осі ординат – кількість досліджених об’єктів в %. Таким чином, отримані результати свідчать про недостатню ефективність використання монотерапії тироксином в запобіганні розвитку гіпотиреоїдної міокардіопатії. Використання комбінованого з кальцитоніном лікування дозволяє попередити маніфестні ураження міокарда, що може слугувати теоретичним підґрунтям для розроблення схем лікування післяопераційного гіпотиреозу. Максимальна збереженість кардіоміоцитів спостерігається при трансплантації фетальних щитовидних залоз, що ставить питання про доцільність подальших пошуків, враховуючи і виявлені негативні аспекти цього виду лікування.
висновки
В дисертації наведено теоретичне узагальнення та нове вирішення актуального питання морфофункціональних змін в міокарді правого передсердя та лівого шлуночка в динаміці формування післяопераційного гіпотиреозу та можливостей їх корекції.
1. Розроблена модель післяопераційного гіпотиреозу на щурах, адекватність якої підтверджена біохімічними, фізіологічними, морфологічними методами.
2. В динаміці розвитку експериментального гіпотиреозу структурні зміни у міокарді як правого передсердя, так і лівого шлуночка носять однотипний характер і спрямованість, але розрізняються за ступенем їх виразності. Встановлено, що у міокарді обох досліджених відділів на ранніх етапах розвиваються реактивні зміни, які передують розвитку компенсаторно-пристосувальних процесів, що в подальшому завершуються декомпенсацією та розвитком хронічної серцево-судинної недостатності. Маніфестність змін в усі досліджені терміни спостережень превалює у міокарді правого передсердя.
3. Реактивні зміни проявляються вираженим гетеротипізмом як кардіоміоцитів, так і інших клітин міокарду. Характерною особливістю міокарда на ранніх строках (14 доба) експерименту була наявність значної кількості апоптозно змінених кардіоміоцитів та ендотеліоцитів.
4. Компенсаторно-пристосувальні механізми найбільшого розвитку набувають на 50 добу після операції та стосуються енергетичного, білоксинтетичного, скоротливого апаратів кардіоміоцитів. Це проявляється гіперплазією та гіпертрофією мітохондрій, канальців зернистої ендоплазматичної сітки, репарацією міофібрил.
5. Збільшення терміну спостережень (100 діб) вказує на зрив компенсації та виникнення хронічних змін у міокарді обох відділів серця, ознаками чого є деструкція міофібрил, внутрішньоклітинних мембран та сарколеми, з виходом вмісту кардіоміоцитів в інтерстицій, мікрогеморагії, міокардіофіброз.
6. Встановлено, що лікування L-тироксином тиреоїдектомованих щурів не попереджує формування структурних та функціональних змін, але відстрочує їх у часі. Гострі альтеративні процеси проявляються через 35 діб після операції, а через 100 діб з’являються перші ознаки зриву компенсації. L-тироксин стимулює розвиток білоксинтетичного апарату кардіоміоцитів, тоді як у енергетичному та скоротливому апаратах він не запобігає деструктивним змінам. Він посилює колагеноутворення у порівнянні з нелікованими тиреоїдектомованими тваринами.
7. Комбіноване лікування L-тироксином та кальцитоніном зменшує глибину дистрофічних явищ у міокарді при відсутності вираженої стадійності перебігу гіпотиреоїдної міокардіопатії та запобігає розвитку міокардіофіброзу.
8. Доведено, що в розвитку гіпотиреоїдної міокардіопатії значну роль відіграють порушення гомеостазу кальцію, що морфологічно проявляється розширенням канальців саркоплазматичної сітки, відкладанням депозитів кальцію у мітохондріальному матриксі, ділянками ущільнення кортикального шару цитоплазми, контрактурами міофібрил
8-09-2015, 22:26