Рис.1. Каріограма пацієнта з синдромом Сміта-Мадженіса.
Стрілкою вказано делецію короткого плеча хромосоми 17 у ділянці p11.2, пунктиром вказано інверсію хромосоми 9. GTG-забарвлення. Об’єктив 100.Каріотип пацієнта: 46,XX,del(17)(p11.2p11.2),inv(9)(p12q12)
Наведені в світовій літературі дані показують, що діагностика синдрому „котячого ока” ускладнена варіабельністю клінічних та цитогенетичних характеристик (Berendsetal., 2001; Meins еtal., 2003). У нашому дослідженні пацієнт не мав колобоми райдужки та преаурікулярних дермоїдних привісків, однак поєднання ВВС та ректо-анальної атрезії, що є характерними для синдрому, дозволило запідозрити трисомію за хромосомою 22. Під час стандартного цитогенетичного аналізу нами було виявлено наявність додаткової хромосоми 22 з делецією в ділянці q11.2. та верифіковано діагноз – синдром „котячого ока”. Враховуючи той факт, що на сьогодні у світі описано лише близько 60 випадків синдрому „котячого ока”, кожний виявлений та підтверджений випадок має свою наукову цінність і є важливим для розширення знань про кореляцію фенотипу-каріотипу та можливості діагностики даного синдрому.
В інших трьох випадках нами виявлено зміни хромосомного матеріалу, які не вирішували питання щодо верифікації діагнозу ССА в зв’язку з тим, що у виявлених структурних перебудовах досліджувані хромосоми 15, 22 участі не брали (2 випадки), або наявність/відсутність критичної ділянки в структурно-перебудованій хромосомі була під сумнівом (1 випадок). Так, у пробанда з підозрою на синдром мікроделеції 22q11.2 виявлено структурну перебудову за участю хромосом 12 та 22, каріотип пацієнта: 46,XX,der(12)t(12;22)(q12;q11.2),-22. Для встановлення походження дериватної хромосоми проведено каріотипування батьків цієї дитини. Каріотип батька: 46,XY, каріотип матері: 46,XX,t(12;22)(q12;q11.2). Критична для розвитку синдрому мікроделеції 22q11.2 ділянка q11.2 була задіяна у реципрокній транслокації, яку виявлено в каріотипі у матері. В той же час в результаті проведення стандартного цитогенетичного аналізу не було можливим достовірно визначити наявність чи відсутність критичної ділянки в дериватній хромосомі пробанда (табл. 2). Це стало підставою для подальшого лабораторного дослідження даного випадку.
Таблиця 2
Порівняння між частковими каріограмами пробанда з підозрою на синдром мікроделеції 22 q 11.2 та його матері.
Пацієнт |
Каріотип |
Гомоло-ги хромосоми 12 | Гомологи хро мосоми 22 |
Структурно перебудовані хромосоми в каріотипі: | |
пробанд |
46,XX,der(12) t(12;22) (q12;q11.2)mat,-22 |
|
|||
| N N | N | der(12)t(12;22)(q12;q11.2) | |||
| мати пробанда | 46,XX,t(12;22) (q12;q11.2) |
|
|||
| N | N | der(12)t(12;22) | der(22)t(12;22) | ||
У іншому випадку за допомогою аналізу хромосом метафазної конденсації у пробанда з підозрою на СПВ виявлено робертсонівську транслокацію між хромосомами 14 та 22. При цитогенетичному аналізі в родині пацієнта встановлено: каріотип матері – 46,XX, каріотип сестри – 46,XX. У батька виявлено таку саму структурну перебудову, як і у пробанда. Дитина мала характерні для СПВ клінічні ознаки, тому наявність структурної перебудови батьківського походження не відігравала визначальної ролі у виникненні та розвитку клінічної картини. В той же час, у дитини не було виявлено делеції критичної ділянки хромосоми 15, що потребувало подальшого аналізу із застосуванням більш чутливих методів.
При дослідженні каріотипу пацієнта з підозрою на синдром мікроделеції 22q11.2 та характерними клінічними ознаками стандартним цитогенетичним методом виявлено інверсію хромосоми 9, яка оцінюється як поліморфізм. Інших змін хромосомного матеріалу на рівні метафаз у каріотипі виявлено не було, тому діагноз на даному етапі дослідження не встановлено.
Всього, за результатами стандартного цитогенетичного аналізу верифіковано діагноз у 7 пацієнтів з 78 (8,97%). Застосування стандартного цитогенетичного методу не дозволило верифікувати діагноз у 71 особи (85,89%), що потребувало подальшого дослідження їхнього каріотипу із використанням чутливіших методів діагностики ХП.
Високочутливий цитогенетичний метод застосованоу 74 осібз метою виявлення мікроделеції в критичних ділянках досліджуваних хромосом 4, 5, 7, 15, 22 та для уточнення точок розривів - з’єднання у структурно перебудованих хромосомах, виявлених під час стандартного цитогенетичного методу.
В результаті проведення аналізу хромосом прометафазної конденсації попередній клінічний діагноз - синдром мікроделеції 22q11.2 підтверджено у трьох пробандів з 31 особи (3,85%), у яких стандартним методом делецію критичної ділянки не було виявлено. З метою запобігання хибно позитивних чи хибно негативних результатів проведено перевірку отриманих даних за допомогою FISH-методу. В одному випадку у пацієнта та у його матері за допомогою високочутливого цитогенетичного методу уточнено точки розривів-з’єднання при структурній перебудові, що не вдалося визначити стандартним цитогенетичним методом: каріотип пробанда після аналізу точок розривів-з’єднання було уточнено з 46,XX,der(12)t(12;22)(q12;q11.2)mat,-22 на 46,XX,der(12)t(12;22)(q13.2;q11.2)mat,-22. В той же час, навіть після застосування високочутливого методу, питання про наявність чи відсутність критичної ділянки q11.2 хромосоми 22 у цього пробанда залишилося відкритим, і даний випадок потребував подальшого дослідження з використанням більш чутливих методів. Для 61 особи (78,21%) остаточний діагноз на даному етапі не встановлено.
Таким чином, використання цитогенетичних (стандартного та високочутливого) методів дозволило верифікувати діагноз у 10 пацієнтів з 78 осіб та виявити батьківське походження дериватних хромосом у двох випадках. Крім того виявлено інші хромосомні перебудови без участі критичних ділянок у трьох пацієнтів. Інформативність цитогенетичних методів для підтвердження попереднього клінічного діагнозу СВХ, синдрому „котячого крику”, ССМ та синдрому „котячого ока” склала 100%, синдрому мікроделеції 22 q11.2 – 3,85%, для клінічних діагнозів СВБ та СПВ – 0%. Загальна інформативність цитогенетичних методів склала 12,82%.
Молекулярно-цитогенетичне дослідження проведено у 71 із 78 пацієнтів, які мали попередній клінічний діагноз ССА (за винятком 7 пацієнтів, яким діагноз було верифіковано за допомогою стандартного цитогенетичного методу). За допомогою FISH-методу вперше в Україні нами підтверджено клінічний діагноз у 18 осіб із 21 пацієнта з СВБ (86%), у 9 осіб із 19 пацієнтів з СПВ (47%) та у 13 осіб із 31 пацієнта з синдромом мікроделеції 22q11.2 (42%).
Застосування FISH-методу дозволило спростувати попередній клінічний діагноз у трьох із 21 пацієнта з СВБ (14%) і у 18 осіб із 31 обстеженого пацієнта з синдромом мікроделеції 22q11.2 (58%).![]()
![]()
В зв’язку з тим, що до розвитку СПВ можуть призводити різні етіологічні чинники, а саме: делеція, мікроделеція в критичній ділянці, функціональна делеція або уніпарентна дисомія та мутація в центрі імпринтингу, навіть наявність двох сигналів локус-специфічного зонду у критичній ділянці 15q11.2-q13 не дозволила спростувати попередній клінічний діагноз. Тому, у 10 осіб із 19 пацієнтів з СПВ остаточний діагноз не було верифіковано (53%).
Отже, при дослідженні каріотипу 71 пацієнта з підозрою на ССА FISH-методом діагноз підтверджено у 40 пацієнтів (51,28%) та спростовано у 21 пацієнта (26,92%). Діагноз не верифіковано у 10 пробандів (12,82%). Таким чином рівень інформативності застосування молекулярно-цитогенетичного методу в нашому дослідженні склав 78,2%.
Важливо відзначити, що із 71 пацієнта для чотирьох (два пробанди з підозрою на СВБ та два пробанди з підозрою на синдром мікроделеції 22) діагноз підтверджено при молекулярно-цитогенетичному дослідженні на інтерфазних ядрах. Така необхідність виникла внаслідок низького мітотичного індексу у цих хворих. На сьогодні, інтерфазний метод посідає значне місце у виявленні ХП при проведенні пренатальної діагностики (Ворсанова, 2006). Наші дані показують, що цей метод може бути методом вибору при необхідності швидкої верифікації діагнозу ССА. Однак, після встановлення або спростування діагнозу на інтерфазних ядрах, обов’язковим є проведення стандартного цитогенетичного аналізу для перевірки наявності інших змін хромосомного набору, що і було нами вище показано.
В зв’язку з тим, що у 10 пацієнтів з попереднім клінічним діагнозом СПВ діагноз не верифіковано, ДНК пацієнтів та їхніх батьків було відправлено на молекулярний аналіз. В результаті проведення аналізу метилування ДНК та мікросателітного аналізу у трьох пацієнтів діагноз СПВ підтверджено, у двох – спростовано. Для 5-ти пацієнтів результати молекулярно-генетичного обстеження виявились не інформативними (6,4%).
Аналіз даних, отриманих при проведенні комплексного дослідження пацієнтів з підозрою на ССА із залученням стандартного та високочутливого цитогенетичних методів, FISH-аналізу та молекулярного аналізу показав, що процес підтвердження або спростування попереднього клінічного діагнозу є складним та охоплює декілька етапів. Комплексне застосування цитогенетичних, молекулярно-цитогенетичних та молекулярних методів дозволило верифікувати діагноз у 93,6% пацієнтів з підозрою на ССА.
У відповідності з одержаними результатами, нами було створено загальний алгоритм лабораторного генетичного обстеження пацієнтів з підозроюна ССА (рис. 2) та розроблено диференційно-діагностичний підхід з цілеспрямованим використанням лабораторних генетичних методів для кожного нозологічного випадку.
Алгоритм лабораторного генетичного обстеження пацієнтів з підозрою на ССА
Перший етап лабораторного генетичного обстеження – цитогенетичний аналіз .
Усім пацієнтам із попереднім клінічним діагнозом ССА проводять стандартне цитогенетичне дослідження.
· У разі виявлення делеції/дуплікації критичних ділянок хромосом 4, 5, 7, 15, 17, 22 або їхньої участі у структурних перебудовах з іншими хромосомами з втратою критичної ділянки діагноз підтверджується. Пацієнт та члени його родини направляються для завершення медико-генетичного консультування до лікаря-генетика.
· При виявленні інших структурних перебудов, у яких критична ділянка не задіяна та при необхідності встановлення точок розривів-з’єднання можливим є використання високочутливого цитогенетичного аналізу.
· Виявлення структурних перебудов у каріотипі дитини обумовлює необхідність каріотипування батьків дитини, що дозволяє встановити батьківське походження перебудованих хромосом та уточнити точки розривів у хромосомах, які брали участь у структурній перебудові.
· При відсутності метафазних пластинок на хромосомних препаратах одразу переходять до другого етапу –використовуютьFISH-аналіз на інтерфазних ядрах.
Варто зазначити, що для пацієнтів, у яких діагноз був підтверджений за допомогою FISH-методу на інтерфазних ядрах, у подальшому ми рекомендуємо провести стандартний цитогенетичний аналіз із метою виключення інших структурних перебудов у каріотипі, оскільки вони можуть впливати на стан дитини.
При характерній клінічній картині ССА і нормальному каріотипі переходять до
другого етапу – молекулярно-цитогенетичної діагностики .
· FISH-діагностику проводять усім пацієнтам з попереднім клінічним діагнозом ССА, у яких на першому етапі не виявлено делецію критичної ділянки досліджуваної хромосоми.
· При виявленні мікроделеції критичної ділянки діагноз підтверджується, і родина повертається до лікаря-генетика для подальшої консультації.
· Якщо мікроделецію критичної ділянки не виявлено, діагноз спростовується, і пацієнт направляється до лікаря-генетика. У цьому випадку найвірогідніше, що
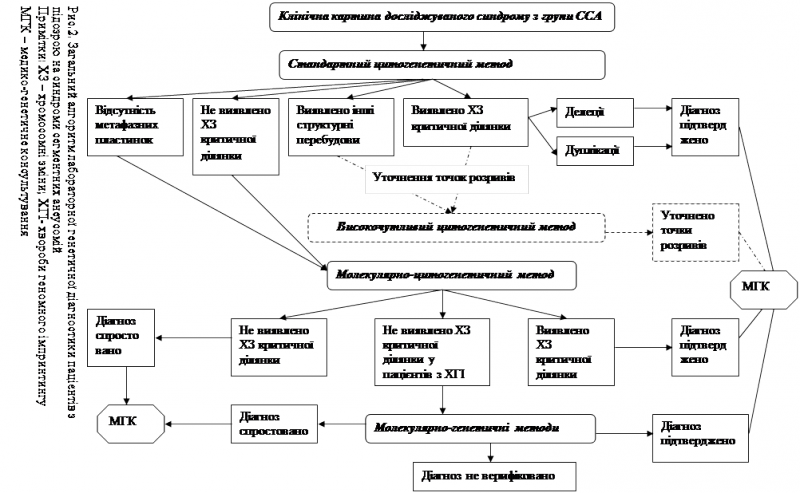
пацієнт має патологію іншого генезу і тому потребує подальшої диференційної діагностики.
· Винятком із цього правила є пацієнти з хворобами геномного імпринтингу (наприклад, синдром Прадера-Віллі). Якщо під час проведення FISH-аналізу не виявлено мікроделеції в каріотипі пробанда з попереднім клінічним діагнозом СПВ, це є приводом для подальшого дослідження із застосуванням методів молекулярного аналізу з метою виявлення функціональної делеції, уніпарентної дисомії або мутації центру імпринтингу. Відповідно з результатами молекулярного аналізу у таких випадках діагноз або підтверджується або спростовується, що є приводом для диференційної діагностики. Молекулярне дослідження також рекомендовано при необхідності визначення батьківського походження мікроделеції, виявленої на попередніх етапах обстеження та при плануванні наступної вагітності в родині, яка має дитину з ССА.
Отже, розроблений нами алгоритм дозволяє вибрати оптимальні шляхи проведення генетичного обстеження пацієнтів з підозрою на ССА та може бути використаний для визначення подальшої стратегії медико-генетичного консультування родин, що мають дитину з ССА.
На основі одержаних результатів та враховуючи рекомендований нами алгоритм, було розроблено диференційно-діагностичний підхід до формування груп пацієнтів з підозрою на ССА з метою індивідуалізованого та цілеспрямованого використання цитогенетичних, молекулярно-цитогенетичного та молекулярно-генетичного методів. З 78 обстежених пацієнтів ми сформували три групи. До першої групи увійшли пацієнти з наступними синдромами: Вольфа-Хіршхорна, „котячого крику”, Сміта-Мадженіса та „котячого ока”. При діагностиці цих нозологічних форм ХП економічно та стратегічно виправданим є застосовування стандартного цитогенетичного методу. У разі, якщо делецію/дуплікацію не було виявлено, рекомендуємо одразу застосовувати молекулярно-цитогенетичний метод з використанням локус-специфічних зондів на відповідні критичні ділянки. Високочутливий цитогенетичний метод у даної групи пацієнтів може бути застосований при необхідності уточнення точок розривів-з’єднання хромосомного матеріалу, визначеного при стандартному методі.
До другої групи увійшли пацієнти з підозрою на синдром Вільямса-Бойрена та синдром мікроделеції 22q11.2. Для верифікації діагнозу у пацієнтів з цієї групи ми рекомендуємо одночасне застосування стандартного цитогенетичного та молекулярно-цитогенетичного методів діагностики ХП. Такий підхід дає можливість прискорити верифікацію діагнозу, що найбільш важливо при необхідності термінового кардіо-хірургічного втручання.
До окремої третьої групи увійшли пацієнти з підозрою на синдром Прадера-Віллі, для яких верифікація діагнозу є складною, багатоступеневою і потребує поступового залучення використаних нами методів діагностики. Для встановлення остаточного діагнозу ми рекомендуємо одночасно застосувати стандартний цитогенетичний та молекулярно-цитогенетичний методи діагностики на першому етапі верифікації діагнозу СПВ, та, якщо делецію/мікроделецію критичної ділянки хромосоми 15 не виявлено, продовжити дослідження із використанням методів молекулярного аналізу.
ВИСНОВКИ
В дисертаційній роботі вперше запропоновано новий підхід щодо оптимізації генетичної діагностики ССА, який базується на визначенні інформативності стандартних та високочутливих цитогенетичних і молекулярно-цитогенетичного методів та створенні алгоритму генетичного обстеження пацієнтів з підозрою на ССА. Показно гетерогенність прояву кожного із досліджених синдромів як за клінічними, так і за цитогенетичними характеристиками, і необхідність застосування комплексу лабораторних генетичних підходів для підтвердження або спростування попереднього клінічного діагнозу.
1. Доведено необхідність застосування стандартного цитогенетичного методу на першому етапі верифікації діагнозу ССА, який дозволив підтвердити попередній клінічний діагноз ССА у 8,97% пацієнтів, виявити інші хромосомні перебудови у 3,8% пацієнтів, встановити додатковий діагноз – синдром трисомії за довгим і, частково, коротким плечем хромосоми 12, який не відноситься до групи ССА. Встановлено батьківське походження дериватних хромосом у 33,3% випадків та обгрунтовано необхідність цитогенетичного аналізу у батьків при виявленні змін каріотипу у їхньої дитини.
2. Показано, що застосування високочутливого цитогенетичного методу дозволяє виявляти мікроделеції (3,8%) та конкретизувати результати стандартного цитогенетичного аналізу і точки розривів при структурних перебудовах хромосом.
3. Визначено високу специфічність FISH-методу для верифікації діагнозу ССА, застосування якого дозволило підтвердити попередній клінічний діагноз у 86% пацієнтів з синдромом Вільямса-Бойрена, у 47% – з синдромом Прадера-Віллі та 42% пацієнтів з синдромом мікроделеції 22q11.2; спростувати діагноз у 14% пацієнтів з синдромом Вільямса-Бойрена та 58% пацієнтів з синдромом мікроделеції 22q11.2, а також провести диференційну діагностику між синдромом Вільямса-Бойрена (МІМ 194050) та спадковим судинним захворюванням – ізольованим надклапанним стенозом аорти (МІМ 185500).
4. Вперше в Україні та вдруге в світі виявлено поєднання у одного пацієнта двох синдромів: синдрому мікроделеції 22q11.2 та синдрому трисомії за довгим і, частково, коротким плечем хромосоми 12. За допомогою поетапного застосування стандартного цитогенетичного методу та FISH-методу обгрунтовано доцільність одночасного використання обох методів при підозрі на ССА та виявленні інших структурних перебудов в каріотипі пробанда.
5. Встановлено високу інформативність FISH-аналізу із застосуванням локус-специфічних ДНК-зондів на інтерфазних ядрах при відсутності мітотичних хромосом на препаратах лімфоцитів периферичної крові (5,1%).
6. Визначено, що рівень інформативності застосування стандартного цитогенетичного та високочутливого цитогенетичного методів для підтвердження попереднього клінічного діагнозу синдромів: Вольфа-Хіршхорна, „котячого крику”, Сміта-Мадженіса та „котячого ока” склав 100%, синдрому мікроделеції 22q11.2 – 3,85%, в той же час як для синдромів Вільямса-Бойрена та Прадера-Віллі – 0%. Загальна інформативність цитогенетичних методів в нашому дослідженні склала 12,8%.
7. Встановлено, що загальна інформативність молекулярно-цитогенетичного методу для підтвердження або спростування попереднього клінічного діагнозу ССА в нашому дослідженні склала 78,2%, що обґрунтовує доцільність використання FISH-аналізу для верифікації діагнозу синдромів з групи ССА.
8. Визначено, що застосування цитогенетичних, молекулярно-цитогенетичних та молекулярних методів дозволило верифікувати діагноз у 93,6% пацієнтів з підозрою на ССА, на підставі чого розроблено комплексний підхід щодо оптимізації індивідуалізованої діагностики кожного синдрому з групи ССА, який включає формування диференційно-діагностичних груп пацієнтів та створення алгоритму генетичного обстеження пробандів та їх родичів.
Практичні рекомендації
Для лікарів-генетиків, неонатологів, педіатрів, кардіологів, кардіохірургів, дитячих неврологів, імунологів:
1. Направляти на цитогенетичний та молекулярно-цитогенетичний аналіз пацієнтів з характерними для синдромів з групи ССА вродженими вадами розвитку та вродженими вадами серця з метою підтвердження/спростування попереднього клінічного діагнозу.
2. Верифікацію діагнозу у дітей із підозрою на ССА доцільно проводити за запропонованим алгоритмом генетичного обстеження пацієнтів з даною патологією.
3. Виявлення у пробанда структурних перебудов хромосомного матеріалу за участю критичних ділянок хромосом 4, 5, 7, 15, 17, 22 або за участю інших хромосом є підставою для цитогенетичного дослідження лімфоцитів периферійної крові його батьків.
4. У всіх пацієнтів з надклапанним стенозом аорти рекомендовано проводити FISH-аналіз з метою підтвердження або спростування синдрому Вільямса-Бойрена.
5. Виявлення у плоду під час пренатального УЗД вад серця, що характерні для ССА обґрунтовує необхідність проведення пренатальної молекулярно-цитогенетичної діагностики плоду.
6. Наявність мікроделеції/мікродуплікації у пробанда є підставою для проведення пренатальної діагностики в родинах, що мають дитину з певним синдромом з групи ССА.
ПЕРЕЛІК НАУКОВИХ ПРАЦЬ, ОПУБЛІКОВАНИХ ЗА ТЕМОЮ ДИСЕРТАЦІЇ
1. Горовенко Н.Г., Євсеєнкова О.Г., Зерова-Любимова Т.Е., Тищенко Н.О. Роль молекулярно - цитогенетичного методу в діагностиці синдрому Вільямса-Бойрена // Вісник Українського товариства генетиків і селекціонерів. – 2006. – Т.4, №2. – С.
8-09-2015, 19:29